V


ALMAAREFA UNIVERSITY
CLINICAL APPROACH & MANAGEMENT OF ANAEMIA CBD
Dr Hisham M Abdelrahim
LEARNING OBJECTIVES
- To define anaemia, classification and the clinical features of each type.
- To describe the investigations needed to diagnose anemia.
- To outline the management of the common types of anemia.
Introduction
-
Anemia is defined as a decrease in the quantity of circulating red blood cells (RBC), represented by a reduction in: ① hemoglobin concentration (Hb)², hematocrit (Hct), or RBC count.
-
It is a common condition that can be caused by:
-
inadequate RBC production, by the BM excessive RBC destruction, or blood loss.
-
The most common cause is iron deficiency.
WHO definition of anemia:
- in Females: Hb level < 12g/dl
- in males: Hb level < 13g/dl
CONT-
-
Clinical features, if present, are mostly nonspecific and may include: fatigue, dyspnea, conjunctival pallor, tachycardia and systolic flow murmur.
-
Once anemia has been established, the mean corpuscular volume (MCV) should be checked to distinguish between microcytic, normocytic, and macrocytic anemia and to determine the next diagnostic steps.
-
Reticulocyte count can also be used to evaluate the bone marrow response.
(1) The fact of the
Severity
☐ Mild anemia > Hb level: below normal range based on age and sex, but > 10–11 g/dL > Presentation: usually asymptomatic; clinical features of anemia may be present
☐ Moderate anemia > Hb level: ~ 7–10.9 g/dL > Presentation: clinical features of anemia may or may not be present.
☐ Severe anemia > Hb level: ~ 7–8 g/dLo > Presentation: clinical features of anemia typically present; hemodynamic compromise may be present.
Clinical features of anemia
- Asymptomatic
- Exertional dyspnea and fatigue
- Worsening of angina pectoris → in >50 years old → occurs even if have good supply of blood but Hb content is not enough to produce enough oxygenation to the tissue
- Pallor (e.g., on mucous membranes, conjunctivae)
- Growth impairment (chronic anemia) in paediatrics and adolescent.
ok in case there is patient with ischemic heart disease and present with angina, if there is no explanation to why the angina is getting worst, explore for anemia.
(these patients are taking antiplatelet (aspirin) and it could be a source of blood loss in the GIT)
Signs
- Pallor.
- Tachycardia.
- Systolic flow murmur.
- Cardiac failure. → (anemia itself will not cause HF, but it makes HF easier to happen)
- if there is already an underlying pathology in the heart muscle
- Specific signs seen in the different types of anaemia include:
- koilonychia – spoon-shaped nails seen in longstanding iron deficiency anaemia (Pathognomonic feature for iron deficiency anemia)
- jaundice – found in haemolytic anaemia
- bone deformities – found in thalassaemia major → There is frontal bossing
- leg ulcers – occur in association with sickle cell disease.
- Anaemia is not a final diagnosis and a cause should always be sought.
- eg. - Peptic ulcer
-
- Ca Colon
-
- Dysfunctional uterine bleeding
- (synthesis of blood is in BM esp. in SKull, Sternum, Vertebrae, so a bone deformity of bossing, or active production, indicates long standing anemia, maybe even since birth eg. Thalassemia)
- because should treat the underlying cause not the anemia symptoms
Accompanying features
- Vary depending on underlying etiology
- Clinical features of shock (if anemia is acute)
- Features of IDA: pica (craving for ice or dirt), brittle nails, angular cheilitis, atrophic glossitis
- Features of megaloblastic anemia: oral sores, mucosal bleeding, paresthesias, ataxia
- Clinical features of hemolysis → Jaundice, splenomegaly, leg ulcers
- Signs of hemorrhage: e.g., GI bleeding, menorrhagia. ←in the form of:
Features of hyperdynamic state
- Bounding pulses (Collapsed Pulse)
- Tachycardia/palpitations
- Flow murmur
- Pulsatile sound in the ear
- Possibly heart failure (anemia-induced heart failure)
Features of extramedullary hematopoiesis
( blood produced outside BM) (alternative site for synthesis of RBC)
- Features of extramedullary hematopoiesis may be present in certain severe, chronic forms of anemia (e.g., thalassemia, myelofibrosis).
- Hepatosplenomegaly → very large
- Paravertebral mass
- Widening of diploic spaces of the skull
Other classifications of anemia
- Time course: acute vs. chronic
- Inheritance: inherited vs. acquired
- Etiology: primary vs. secondary
- RBC proliferation: hypoproliferative (decreased RBC production) vs. hyperproliferative (increased RBC destruction or blood loss)
- (bleeding)
e.g. Sickle Cell, Thalassemia, G6PD e.g. Peptic ulcer causing iron deficiency anemia e.g. Secondary to connective tissue disease like RA or SLE. e.g. - BM failure
- red cell aplasia e.g. - red cell aplasia
Normal indices
| Box 16.1 Normal values for peripheral blood | ||
|---|---|---|
| Male | Female | |
| Haemoglobin (Hb) (g/L) | 135–175 | 115–155 |
| Packed cell volume (PCV) (haematocrit; L/L) | 0.4–0.54 | 0.37–0.47 |
| Red cell count (RCC) (10¹²/L) | 4.5–6.0 | 3.9–5.0 |
| Mean corpuscular volume of red cell (MCV) (fL) | 80–96 | |
| Mean corpuscular haemoglobin (MCH) (pg) | 27–32 | |
| Mean corpuscular haemoglobin concentration (MCHC) (g/L) | 320–360 | |
| Red blood cell distribution width (RDW) (%) | 11–15 | |
| White cell count (WCC) (10⁹/L) | 4.0–11.0 | |
| Platelets (10⁹/L) | 150–400 | |
| Erythrocyte sedimentation rate (ESR) (mm/h) | <20 | |
| Reticulocytes | 0.5–2.5% (50–100×10⁹/L) |
Peripheral blood picture
This is used now → blood injected in machine and machine does calculations → investigation needed to make a diagnosis is not used as much now → always needed in addition to automated cell counters.
-
Automated cell counters are used to measure the haemoglobin (Hb) concentration and the number and size of red cells, white cells and platelets. Other indices can be derived from these values.
-
A blood film examined by a haematologist or technician experienced in blood cell morphology is still an essential adjunct to the above, as definitive abnormalities of cells can be seen. → peripheral blood picture can diagnose
-
This is particularly important in some haematological emergencies such as acute promyelocytic leukaemia or thrombotic thrombocytopenic purpura, which require specific therapy within hours of diagnosis to prevent high rates of mortality. → if not treated immediately will get hyperviscosity syndrome and can die within hours
- if Patient has combined iron and folate deficiency, eg. if have malabsorption due to celial disease - so will have features of macrocytic and microcytic anemia. So indices will be normal (normocytic anemia) so make differentiation by doing peripheral blood picture → will see 2 groups of cells (dimorphic picture) → 1. hypochromic microcytic cell
- hypo chromic macrocytic cell
Other indices
- The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia.
- The red cell distribution width (RDW) is a measure of the variability in size of red blood cells. An elevated RDW suggests increased variation in red cell size – that is, anisocytosis – and this is seen in iron deficiency.
- The white cell count (WCC) (or white blood count, WBC) gives the total number of circulating leucocytes in addition to differential counts, specifically enumerating neutrophils, monocytes, lymphocytes, eosinophils and basophils.
- dec Hb, WBC, and platelets → indicate problem in BM (BM failing to produce all components of blood)
Reticulocytes
-
They are young red cells and usually comprise less than 2.5% of red cells. The reticulocyte count gives a guide to erythroid activity in the bone marrow.
-
An increased count is seen with increased marrow activity: for example, following haemorrhage or haemolysis, and during the response to treatment with a specific haematinic.
-
A low count in the presence of anaemia indicates an inappropriate response by the bone marrow and may be seen in bone marrow failure (from whatever cause) or a deficiency of a haematinic. → So not enough source to produce RBCs
could be induced by virus (iron, folate, vit B12)
The erythrocyte sedimentation rate (ESR)
→ indicator of underlying disease when there is rouleaux formation, when there is aggregation of RBCs
- The ESR is the rate of fall of red cells in a column of blood and is a measure of the acute phase response.
- The pathological process may be immunological, infective, ischaemic, malignant or traumatic.
- A raised ESR reflects an increase in the plasma concentration of large proteins, such as fibrinogen and immunoglobulins.
- These proteins cause rouleaux formation, with red cells clumping together and therefore falling more rapidly.
- The ESR increases with age, and is higher in females than in males.
C-reactive protein (CRP)
is indicator for underlying infection→by any microorganism or due to any Rheumatological or immunological disorder
- CRP is a protein produced in the acute phase response.
- It is synthesized exclusively in the liver and rises within 6 hours of an acute event.
- The CRP level rises with fever (possibly triggered by IL-1, IL-6, TNF-α and other cytokines), in inflammatory conditions and after trauma.
- It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of haemoglobin.
*So if want to know if Patient is having infection or active process of Cytokine response to a Pathology, its better to see CRP
Erythropoiesis
- Red cell precursors pass through several stages in the bone marrow.
- The earliest morphologically recognizable cells are proerythroblasts. Smaller erythroblasts result from cell divisions, and precursors at each stage progressively contain less RNA and more haemoglobin in the cytoplasm.
- The nucleus becomes more condensed and is eventually extruded from the late normoblast in the bone marrow when the cell becomes a reticulocyte.
Erythropoiesis
- Reticulocytes contain residual ribosomal RNA and are still able to synthesize haemoglobin. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days.
- Mature red cells are non-nucleated biconcave discs. Nucleated red cells (late erythroblasts) are not normally present in peripheral blood, but are present in extramedullary haemopoiesis and some marrow disorders.
- About 10% of erythroblasts die in the bone marrow, even during normal erythropoiesis. The natural 10% excess production of erythroblasts is known as ineffective erythropoiesis.
- Ineffective erythropoiesis is substantially increased in some anaemias, such as thalassaemia major and megaloblastic anaemia.
(Erythropoietin)EPO
- EPO is a hormone that controls erythropoiesis.
- This heavily glycosylated polypeptide with a molecular weight of 30400 Da is produced in the peritubular cells in the kidneys (90%) and in the liver (10%).
- Its production is regulated mainly by tissue oxygen tension, increasing production in the face of hypoxia: e.g. with anaemia or cardiac or pulmonary disease.
Haemoglobin synthesis
- HbA comprises about 97% of the haemoglobin in adults.
- Two other haemoglobin types, HbA2 (α2δ2) and HbF (α2γ2), are found in adults in small amounts (1.5–3.2% and <1%, respectively).
- Haem synthesis occurs in the mitochondria of the developing red cell.
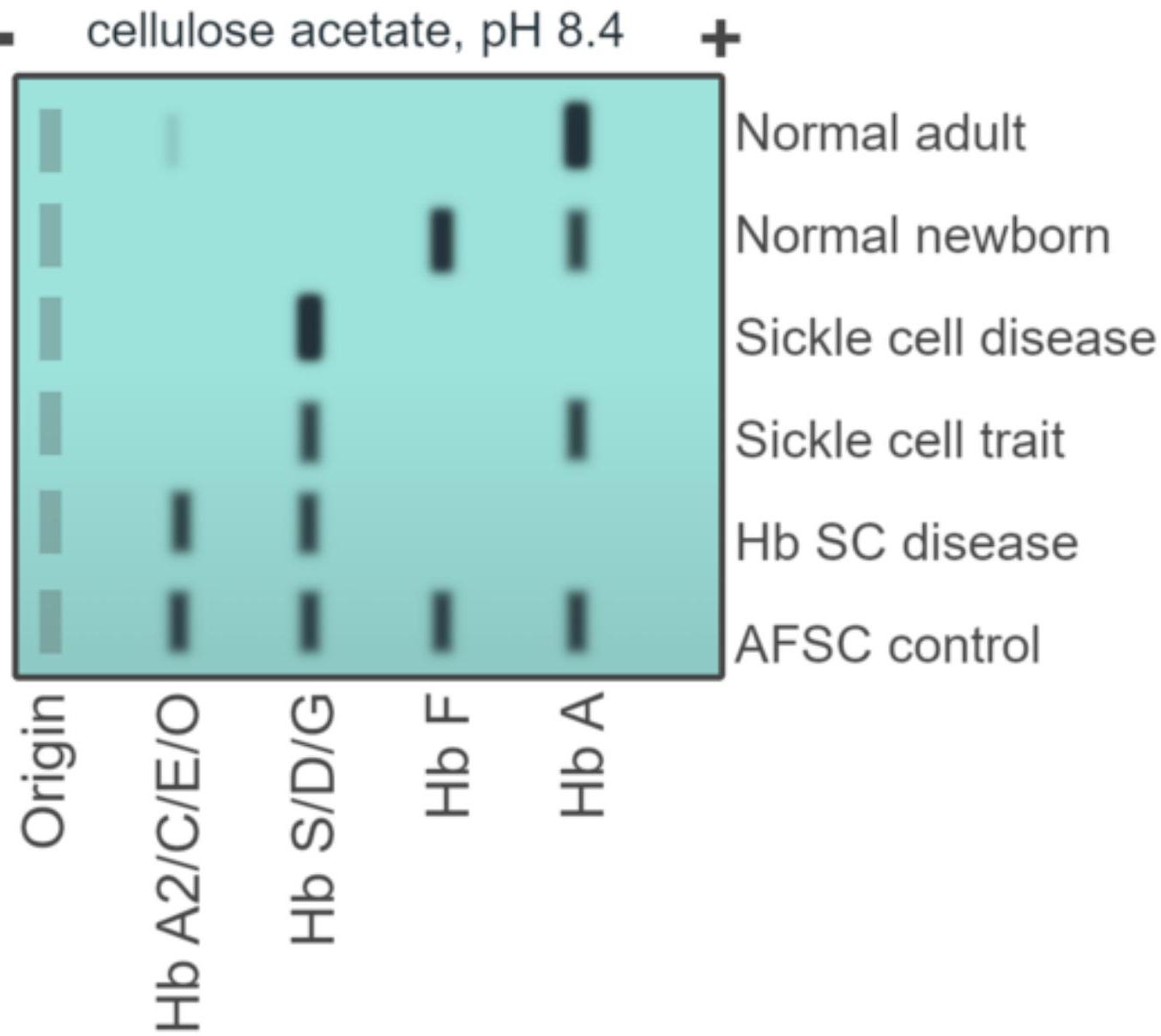
Hemoglobin electrophoresis
- Hemoglobin electrophoresis is a blood test that can detect different types of hemoglobin.
- The test can detect hemoglobin S, the form associated with sickle cell disease, as well as other abnormal types of hemoglobin, such as hemoglobin C.
- It can also be used to investigate thalassemias, which are disorders caused by defective hemoglobin production.
Hemoglobin electrophoresis
Hemoglobin electrophoresis
- cellulose acetate, pH 8.4
BM Aspiration
- provides a film that can be examined by microscopy for the morphology of the developing haemopoietic cells.
- The liquid sample obtained at aspiration can also be used for immunophenotyping (assessment of the presence or absence of antigens on the surface of cells), cytogenetic investigations, assessment for molecular markers or microbiological culture).
BM BIOPSY
- The trephine provides a core of bone that is processed as a histological specimen and allows an overall view of the bone marrow architecture, cellularity and presence/absence of abnormal infiltrates.
- The specimen can be used for immunohistochemical investigations.
- The following are assessed: cellularity of the marrow, type of erythropoiesis (e.g. normoblastic or megaloblastic), morphology of all cell lineages, cellularity of the various cell lines, infiltration of the marrow, i.e. presence of non-haemopoietic cells such as cancer cells, iron stores.
CLASSIFICATION OF ANAEMIA
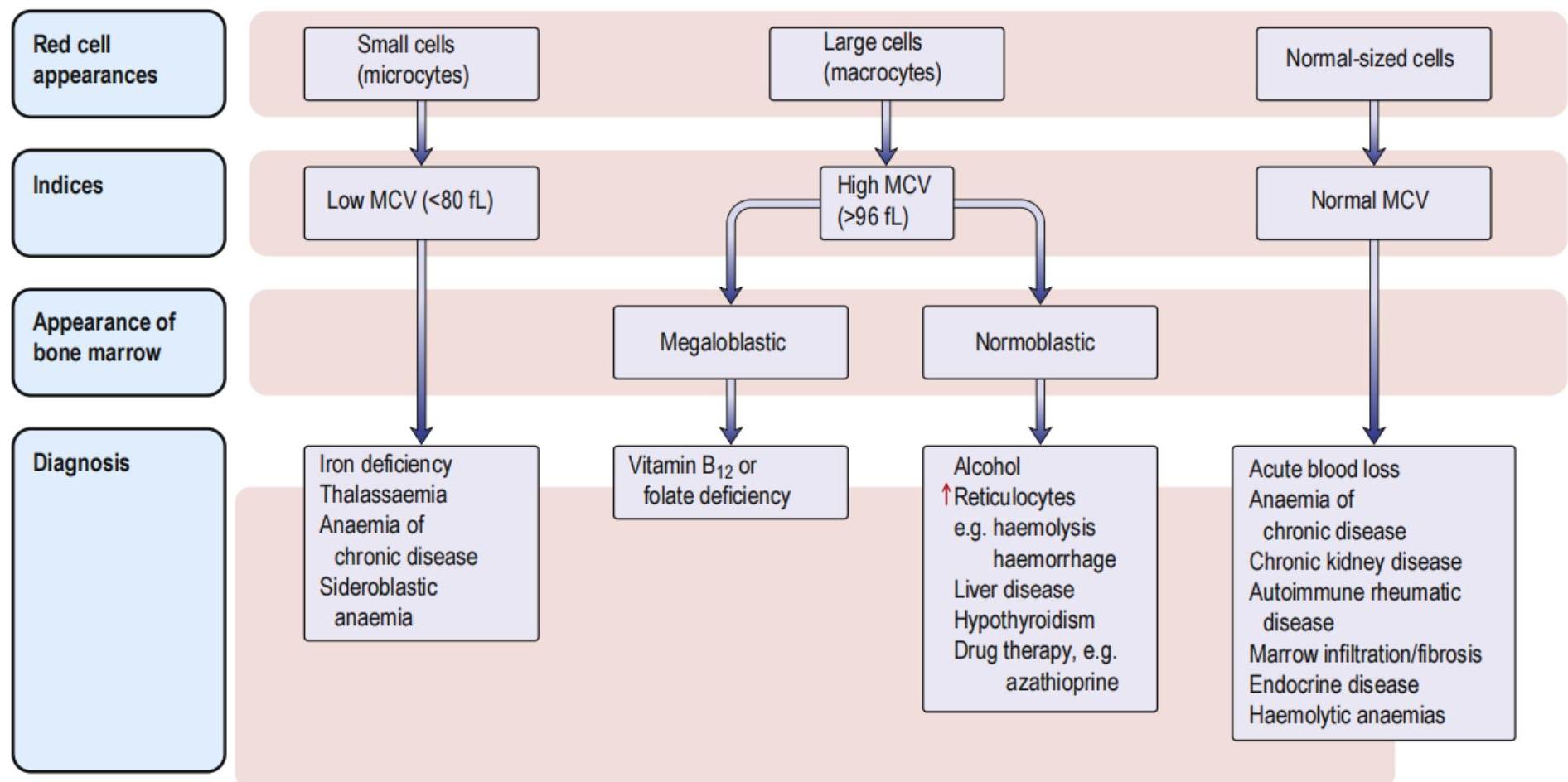 Fig. 16.7 Classification of anaemia. MCV, mean corpuscular volume.
Fig. 16.7 Classification of anaemia. MCV, mean corpuscular volume.
i Box 16.3 Factors influencing iron absorption
- Haem iron is absorbed better than non-haem iron
- Ferrous iron is absorbed better than ferric iron
- Gastric acidity helps to keep iron in the ferrous state and soluble in the upper gut
- Formation of insoluble complexes with phytate or phosphate decreases iron absorption
- Iron absorption is increased with low iron stores and increased erythropoietic activity, e.g. bleeding, haemolysis, high altitude
- Absorption is decreased in iron overload, except in hereditary haemochromatosis, where it is increased
Iron Deficiency Anemia
-
Signs & Symptoms: anemia-related, restless legs, pica, fatigue, glossitis, koilonychia
-
Blood film: microcytosis, hypochromia, pencil cells, targets cells, ↑plt
-
Diagnosis: Ferritin <30 Ferritin >30 + Tsat <20% + inc. TIBC sTFR ↑
-
Treatment:
-
Identify & treat etiology of Fe loss
-
Treat if Ferritin <50 in pregnancy
-
PO
-
Ferrous fumarate: 100 mg Fe
-
Ferrous sulfate: 60 mg Fe
-
Ferrous gluconate: 30 mg Fe
-
Daily vs. q2d
-
Continue for 3 months after ferritin normalizes
-
IV
-
Who?
-
Mod/severe iron def. anemia (Hb <80-100)
-
No response/intolerance to oral
-
Limited time (e.g. pre-op or pregnancy <1 mon from OR/delivery)
-
What?
-
Iron sucrose (e.g. 300 mg IV x 3)
-
Caution: limited evidence in 1st trimester
-
Monoferric (e.g. 1g IV x 1)
-
Caution: little evidence in pregnancy, avoid if known drug all
-
How much?
-
Ganzoni equation to calculate Fe deficit

Distinguishing Microcytic Anemias
- Differential diagnosis
- Iron deficiency
- Thalassemia
- Advanced anemia of chronic disease
- (Sideroblastic anemia; Lead poisoning)
| Fe deficiency | Chronic disease | Thal Trait | |
|---|---|---|---|
| MCV | Low | Normal → Low | Very low (<70) |
| Reticulocytes | Low | Low | High |
| Ferritin | Decreased | Increased | Normal |
| TIBC | Increased | Decreased | - |
| Transferrin saturation | Decreased | Decreased | - |
| Soluble transferrin receptor | Increased | Decreased | - |
| RBC count | Low | Low | High |
| RDW | High | Normal | Normal/ High |
- Thalassemia testing: * β-thal trait: hemoglobin electrophoresis (↑ HbA2)
- α-thal trait: normal electrophoresis; genetic testing
Distinguishing Microcytic Anemias
TABLE 46-2 Interpretation of Iron Studies
| Serum Iron | TIBC | Transferrin Saturation | Ferritin | |
|---|---|---|---|---|
| Iron deficiency | ↓ | ↑ | ↓ | ↓ |
| Anemia of chronic disease | ↓ | N or ↓ | N or ↓ | N or ↑ |
| Thalassemia trait | N | N | N | N or ↑ |
| Sideroblastic anemia | N or ↑ | N or ↑ | N or ↑ | N or ↑ |
N, Normal; TIBC, total iron-binding capacity.
? Box 16.5 Vitamin B₁₂ deficiency and abnormal B₁₂ utilization: further causes
Low dietary intake
- Veganism
Impaired absorption
Stomach
- Pernicious anaemia
- Gastrectomy
- Congenital deficiency of intrinsic factor
Small bowel
-
Ileal disease or resection
-
Bacterial overgrowth
-
Tropical sprue
-
Fish tapeworm (Diphylllobothrium latum)
Abnormal utilization
- Congenital transcobalamin II deficiency
- Nitrous oxide (inactivates B₁₂)
2 Box 16.6 Causes of folate deficiency
Nutritional (major cause)
Poor intake
- Old age
- Poor social conditions
- Starvation
- Alcohol excess (also causes impaired utilization)
Poor intake due to anorexia
- Gastrointestinal disease, e.g. partial gastrectomy, coeliac disease, Crohn’s disease
- Cancer
Antifolate drugs
- Anticonvulsants:
- Phenytoin
- Primidone
- Methotrexate
- Pyrimethamine
- Trimethoprim
Excess utilization
Physiological
- Pregnancy
- Lactation
- Prematurity
Pathological
- Haematological disease with excess red cell production, e.g. haemolysis
- Malignant disease with increased cell turnover
- Inflammatory disease
- Metabolic disease, e.g. homocystinuria
- Haemodialysis or peritoneal dialysis
Malabsorption
- Occurs in small bowel disease but the effect is minor compared with that of anorexia
Vitamin B12 deficiency
Etiology:
- Dietary deficiency (vegans) or meds (PPI, H2 blocker, metformin)
- Absorption issues - no intrinsic factor (pernicious anemia)
- Atrophic gastritis or Pancreatic insufficiency or Terminal ileum disease
Clinical manifestations:
- Anemia-related symptoms, mild jaundice
- Glossitis
- Neurological: subacute combined degeneration (posterior columns & corticospinal tract), peripheral neuropathy, dementia
Hematologic:
- CBC: macrocytic anemia, other lines can be decreased
- film: oval macrocytes, hypersegmented (≥ 5 lobes) neutrophils
Associated Labs:
- low B12, elevated methylmalonic acid & homocysteine levels
- Anti-intrinsic factor and anti-parietal cell antibodies (pernicious anemia)
Treatment:
- Daily high-dose (≥ 1000 μg) oral B₁₂ is as effective as intramuscular supplementation. Hematologic abnormalities should resolve within weeks and neuropsychiatric symptoms within months
- If profound deficiency and need to urgently replace (eg in pregnancy w symptoms) - Vitamin B12 1000 mcg IM daily x7d, then 1000mcg qweek x4, then 1000mcg IM q1month lifelong
- OR once replete, vitamin B12 1000-2000 mcg po OD
Aplastic Anemia (AA)
Definition: stem cell disorder, ↓blood cell production and ↓ marrow cellularity
Presentation: Previously healthy pt with pancytopenia, low reticulocytes, +/- ↑LFTs
Etiology:
- Idiopathic
- Congenital BM failure syndromes
- 2°: pregnancy, autoimmune disease, drugs (NSAIDs, propylthiouracil, carbamazepine)
Diagnosis
- BM biopsy: hypocellular (<25%)
- Severe: ANC <0.5, Plt <20, Retics <60
Treatment
- Required for severe AA
- Supportive transfusion (irradiated), immunosuppression (CsA + anti-thymocyte globulin (ATG) or allohematopoietic stem cell transplant (HSCT)
- If drug-related: may obtain remission with cessation of drug alone
Pancytopenia
| Pancytopenia ddx | |
|---|---|
| Primary | Secondary |
| 1. Paroxysmal Nocturnal Hemoglobinuria (PNH) | 1. Deficiency - B12/Folate, anorexia, ↓T4 |
| 2. Aplastic Anemia | 2. Infection – granulomatous, HIV, sepsis |
| 3. Leukemia | 3. Iatrogenic – chemo, rads, ETOH |
| 4. MDS | 4. Infiltrative – mets, amyloid, Gaucher |
| 5. Myelofibrosis | 5. Hypersplenism |
| Haemolysis | |
|---|---|
| Increased red cell production • ↑ Reticulocytosis/polychromasia • Erythroid hyperplasia of bone marrow | Increased red cell breakdown • Anaemia • ↑ Serum bilirubin • ↑ LDH • ↑ Urobilinogen • +ve urinary haemosiderin • ↓ Plasma haptoglobulin |
| Blood film | |
| Abnormal red cells Haemoglobin electrophoresis Sickle cell disease Hb C, D, E, H DCT +ve DCT -ve Erymidine 5’ nucleotidase ↓ | Fragmentation Microangiopathic haemolytic anaemia, e.g. • Eclampsia • Haemolytic uraemic syndrome • Thrombotic thrombocytopenic purpura • DIC |
| Drugs, e.g. • Methyldopa • Quinine • NSAIDs • Interferon • Ribavarin | Autoimmune haemolytic anaemia IgG – warm IgM – cold |
Hemolytic Anemia
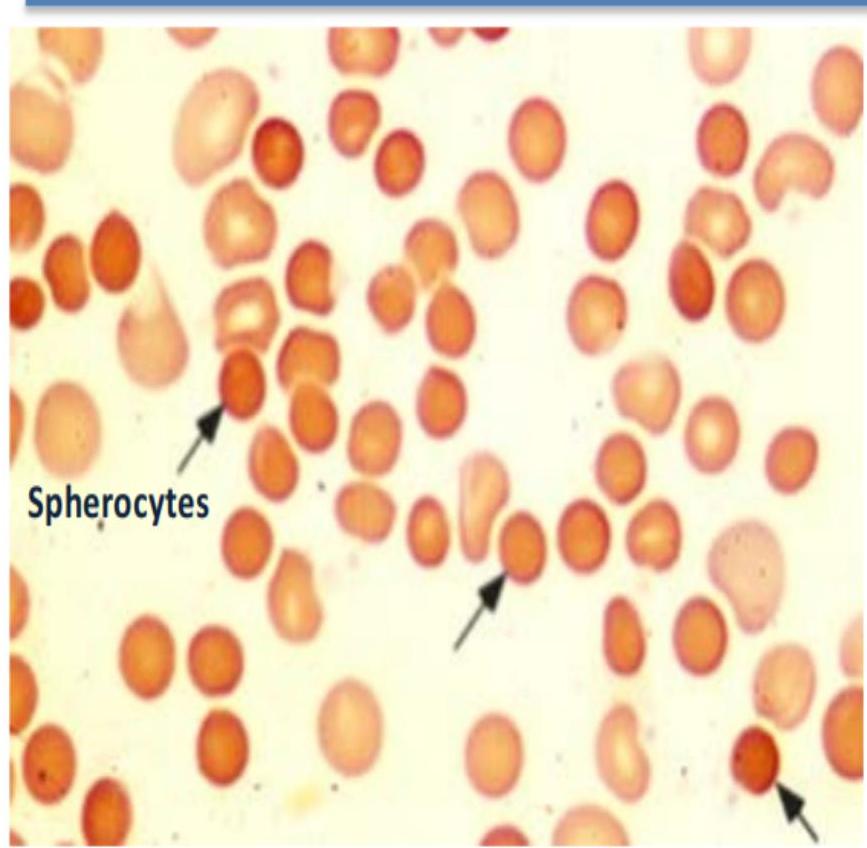
Spherocytes
- Warm autoimmune hemolytic anemia (IgG)
- Hereditary spherocytosis
- Delayed hemolytic transfusion reaction
Agglutination:
- Cold autoimmune hemolytic anemia (IgM)
- Paroxysmal cold hemoglobinuria
Hemolytic Anemia Approach
Based on why the hemolysis occurs
- Intrinsic RBC issues
- Membrane defect: hereditary elliptocytosis / spherocytosis
- Enzyme defect: G6PD deficiency
- Hemoglobinopathy: thalassemia, sickle cell
- Complement-mediated: PNH
- Immune-mediated
- Autoimmune (warm/cold)
- Alloimmune (i.e. hemolytic transfusion reaction)
- Mechanical
- TMA
- Mechanical heart valves
- Other
- Malaria, wilson’s disease
Warm Autoimmune Hemolytic Anemia
IgG-mediated → reticuloendothelial system → hemolysis at body temp
Diagnosis:
- Hemolysis labs
- Blood film: spherocytes
- DAT: +IgG (+/- C3d)
Etiology: 1° vs 2°
- 2°: Lymphoproliferative disease (e.g. CLL), autoimmune (e.g. SLE), drugs (e.g. methyldopa, NSAIDs)
Treatment: 1st line: Prednisone 1 – 1.5 mg/kg/day
2nd line:
- Splenectomy (>8-12 mon)
- Rituximab
Supportive: Folic acid, transfuse for symptoms (requires investigations → call blood bank)
Cold Autoimmune Hemolytic Anemia
IgM fixes complement → intravascular hemolysis at temp < 37°C
Diagnosis:
- Hemolysis labs
- Blood film: agglutination
- DAT +ve C3d (= IgM antibody)
- Thermal amplitude: significant if ≥28°C
Treatment:
- #1: Warm patient!
- Treat underlying cause
- Steroids not helpful
- Rituximab for refractory cases
Etiology: 1° vs 2°
- 2°: Infection (mycoplasma pneumonia, EBV), lymphoproliferative disorder (MGUS, Waldenstroms, lymphoma)
Sickle Cell Disease
- Pathophys: substitution position on Ch11 glutamine to valine → RBC polymerization/sickling when deoxygenated
| Genotype | clinical syndrome | HbA | HbS | Symptoms |
|---|---|---|---|---|
| AA | normal | 97-99% | 0% | - |
| AS | sickle cell trait | 60% | 40% | - |
| SS | sickle cell anemia | 0% | 86-98% | ischemic perfusion hemolysis |
| SC (usually higher baseline Hgb) | hemoglobin SC | HbC 50% | 50% | milder than SS |
Complications by mechanism:
- Anemia/ischemic perfusion injury: VasoOcclusive pain Events (VOE), CVA, ACS, AVN, sequestration, retinopathy, osteoporosis, asplenia
- Hemolysis: priaprism, pulmHTN, ulcers, cholelithiasis Both: nephropathy, VTE, hepatopathy
Pillars of outpatient treatment:
- General medical care: vaccination (asplenia), VOE pain mgmt. plan, complications screening (CBC, retic, hepatic chemistry, renal chemistry, urinalysis, MRI brain, retinopathy screen, TTE, BMD, x-ray joints, PFT)
- Therapies (with approval):
- Hydroxyurea (↓VOC, ↓ACS, ↓transfusions, ↑survival, ↓hospitalizations)
- Exchange transfusion
Sickle Cell Disease
Helpful inpatient Investigations:
- CBC/retic
- Hb electrophoresis
- Chemistries, Troponin, BNP, lactate
- Group and screen – blood bank to phenotype blood (and consider genotyping)
Vasoocclusive crisis:
- Rapid (1hr) analgesia with acetaminophen/NSAID/hydromorphone standing + prn, adjuncts ketamine/regional anesthesia
Acute chest syndrome: clinical diagnosis
- Defn: New infiltrate + new symptom (fever, hypoxemia, tachypnea, chest pain, cough, wheeze)
- Triggers: fluid overload, pulmonary infection, PE, fat embolism, intravascular pulmonary sequestration of sickled erythrocytes
- 50% of sickle cell patients will develop ACS AFTER they were admitted
- Treatment: IVF, analgesia, oxygen, incentive spirometry, Abx, ± transfusion (simple vs. exchange guided by clinical picture and sickle cell parameters)
Complications of sickle cell disease
i Box 16.13 Complications of sickle cell disease requiring inpatient management
-
Pain – uncontrolled by non-opiate analgesia
-
Swollen, painful joints
-
Acute sickle chest syndrome or pneumonia
-
Mesenteric sickling and bowel ischaemia
-
Splenic or hepatic sequestration
-
Central nervous system deficit
-
Cholecystitis (pigment stones)
-
Cardiac arrhythmias
-
Renal papillary necrosis resulting in colic or severe haematuria
-
Hyphaema and retinal detachment
-
Priapism
Transfusion in Sickle Cell Disease
Exchange Transfusion
- Severe acute chest syndrome (multi-organ failure)
- Acute ischemic stroke
- Secondary stroke prophylaxis
- Primary stroke prophylaxis (if high-flow transcranial doppler US)
- Pre-operative if high-risk procedure, Progressive cholestasis, Post-solid organ transplant, High-risk pregnancy, other SCD complications (pulmonary hypertension, priapism, renal disease, ulcers), complications despite top-op transfusion, prevention of pain crises/ACS if failed HU
- Patients needing top-up if Hb >90
Simple (Top Up) Transfusion
- Hemoglobin <50g/L unless from hyperhemolysis
- Mild-moderate acute chest syndrome
- Pre-operative prophylaxis in patients with Hb <90 undergoing low-moderate risk surgery
Beta Thalassemia
Etiology
- Absent or reduced globin gene production
- Epidemiology: Mediterranean, Middle East, Africa, India/Pakistan, SE Asia
- β-thal subtypes: minor/intermedia/major; clinically, more important whether transfusion-dependent
Clinical manifestations/complications
- Ineffective erythropoiesis
- Extramedullary hematopoiesis (skeletal changes, hepatosplenomegaly)
- Hemolysis: anemia, gallstones
- Iron overload: both from ineffective erythropoiesis + transfusion support
Treatment:
- Paradigm moving from major/intermedia to transfusion dependent/non-transfusion-dependent thalassemia; transfusion-dependency based on anemia + complications
- Ferritin measurement q3months, liver/cardiac MRI for iron quantitation
- Iron chelation for features of iron overload (significant toxicities/details of chelators beyond GIM exam)
- Consideration of splenectomy
- Many novel therapeutics on horizon
i Box 16.10 Some types of haemoglobin (Hb)
| Hb | Structure | Comment | |
|---|---|---|---|
| Normal | A | α_{2}β_{2} | Comprises 97% of adult Hb |
| A_{2} | α_{2}δ_{2} | Comprises 2% of adult Hb | |
| Elevated in β-thalassaemia | |||
| F | α_{2}γ_{2} | Normal Hb in fetus from third to ninth month | |
| Increased in β-thalassaemia | |||
| Comprises <1% of Hb in adult | |||
| Abnormal chain production | H | β_{4} | Found in α-thalassaemia |
| Biologically useless | |||
| Barts | γ_{4} | Comprises 100% of Hb in homozygous α-thalassaemia | |
| Biologically useless | |||
| Abnormal chain structure | SC | α_{2}β_{2}Sα_{2}β_{2}C | Substitution of valine for glutamine acid in position 6 of β chain |
| Substitution of lysine for glutamic acid in position 6 of β chain |
Clinical symptoms of thalassaemia
| Thalassaemia carrier | NTDT | TDT |
|---|---|---|
| Thalassaemia trait | ‘Thalassaemia intermedia’ | ‘Thalassaemia major’ |
| No transfusions | Intermittent transfusions | Transfusion dependence |
| Heterozygous for one of: • 1 or 2 deletions of α globin • β0-globin mutation (no β globin produced) • β+-globin mutation (small amount of β globin produced) | HbH disease (3 α gene deletions) • HbE/β-thalassaemia • β-thalassaemia trait with co-inherited multiple copies of α globin • β-thalassaemia major with co-inherited hereditary persistence of fetal haemoglobin or α-thalassaemia | Homozygous or compound heterozygous for either: • β0-globin mutation (no β globin produced) • β+-globin mutation (small amount of β globin produced) |
Fig. 16.19 Clinical syndromes in thalassaemia. The terms ‘thalassaemia intermedia’ and ‘thalassaemia major’ have been replaced by ‘non-transfusion-dependent thalassaemia’ (NTDT) and ‘transfusion-dependent thalassaemia’ (TDT). Patients can fluctuate between these categories, depending on their life circumstances. In addition, the link between the genotype and the phenotype of affected individuals is influenced by modifying factors such as the reactivation of globin or the co-inheritance of changes in the number of -globin genes.
The α thalassaemia
i Box 16.12 The α-thalassaemias
| Number of α-globin genes deleted | Genotype^{a} | Hb type | Clinical picture |
|---|---|---|---|
| 4 | ---/--- | Hb Barts (γ4) | Hydrops fetalis |
| 3 | ---/—α | HbH (β4) | Moderately severe anaemia |
| Splenomegaly | |||
| 2 | —α/—α | HbA | Mild anaemia |
| Or | Some HbH bodies | ||
| ---/αα | |||
| 1 | αα/—α | HbA | Very mild anaemia or no anaemia |
aThe normal α-globin genotype is αα/αα (i.e. four α-globin genes present).
Normal Blood Smear - RBCs
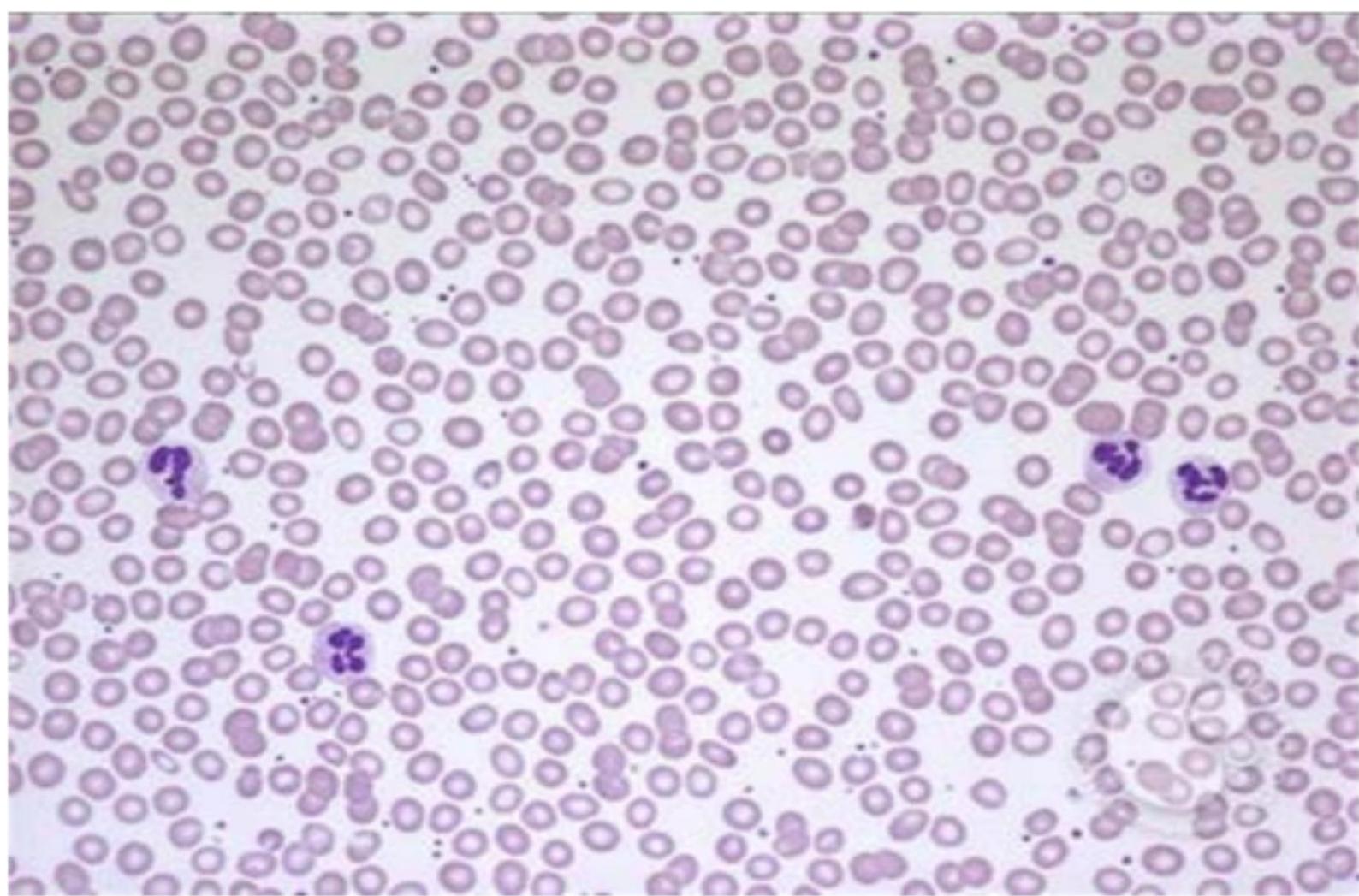
Spherocyte
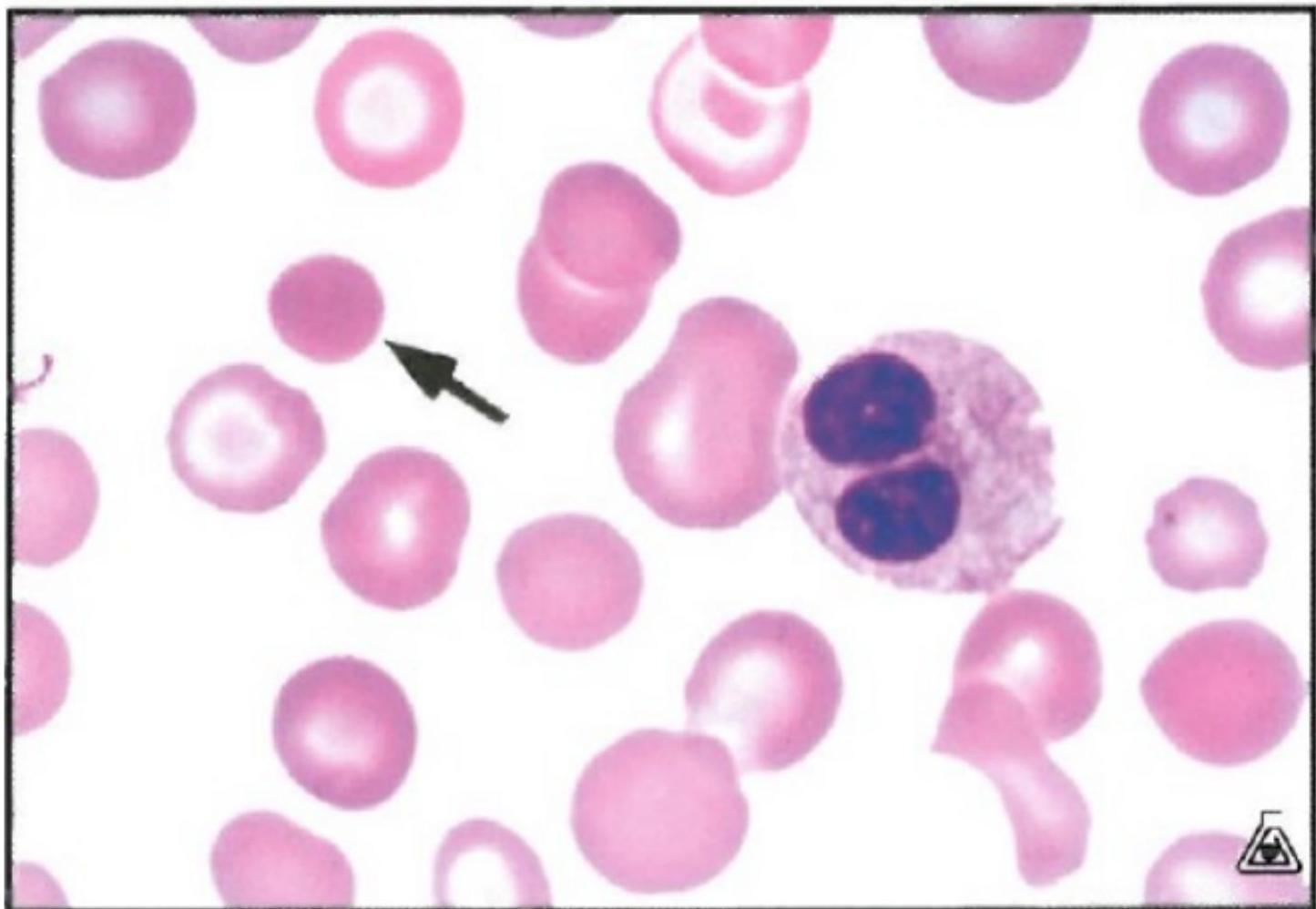
Spherocytes lack central pallor and are smaller than normal RBC. Think immune-mediate hemolysis (e.g. auto or allo immune)
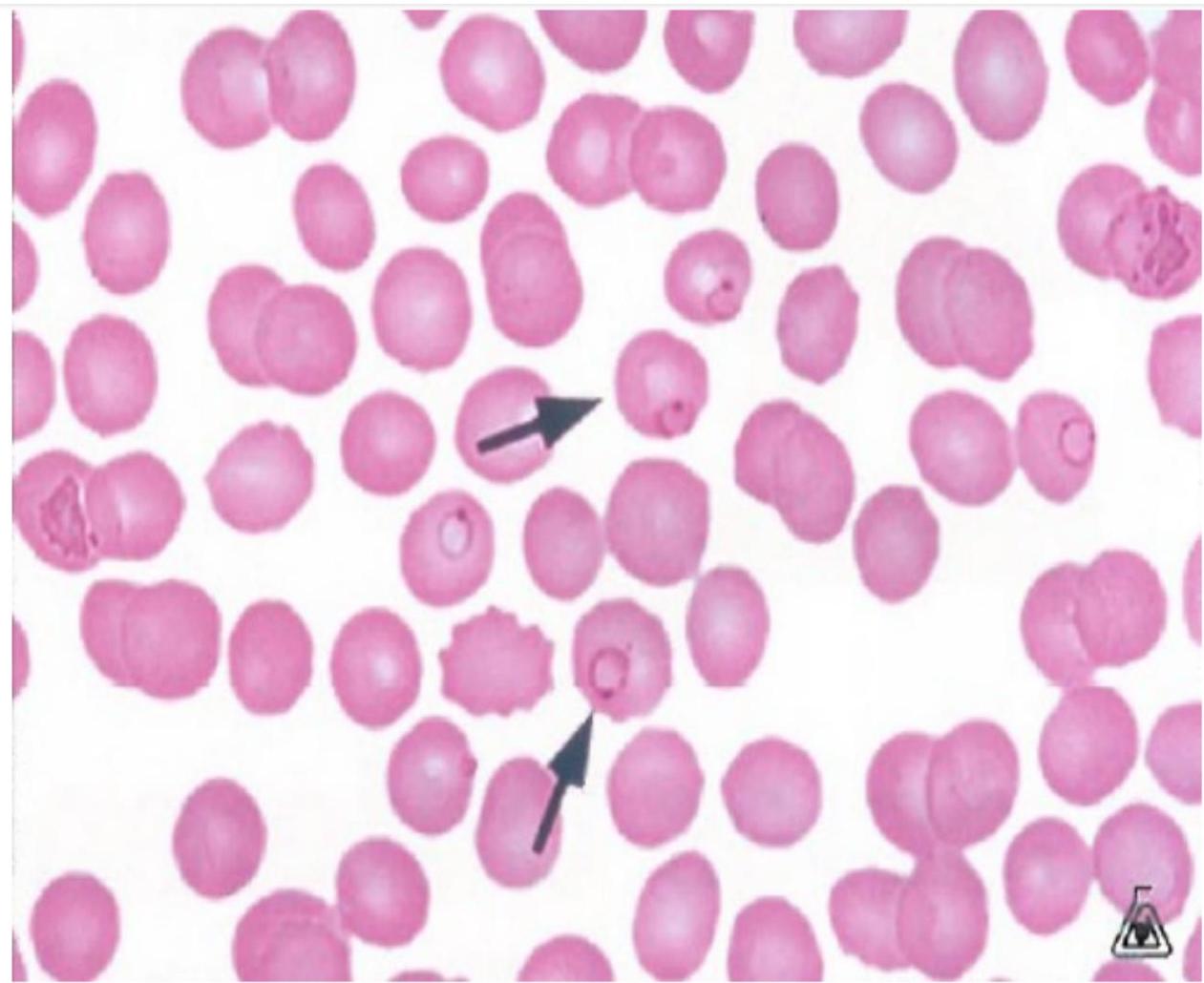
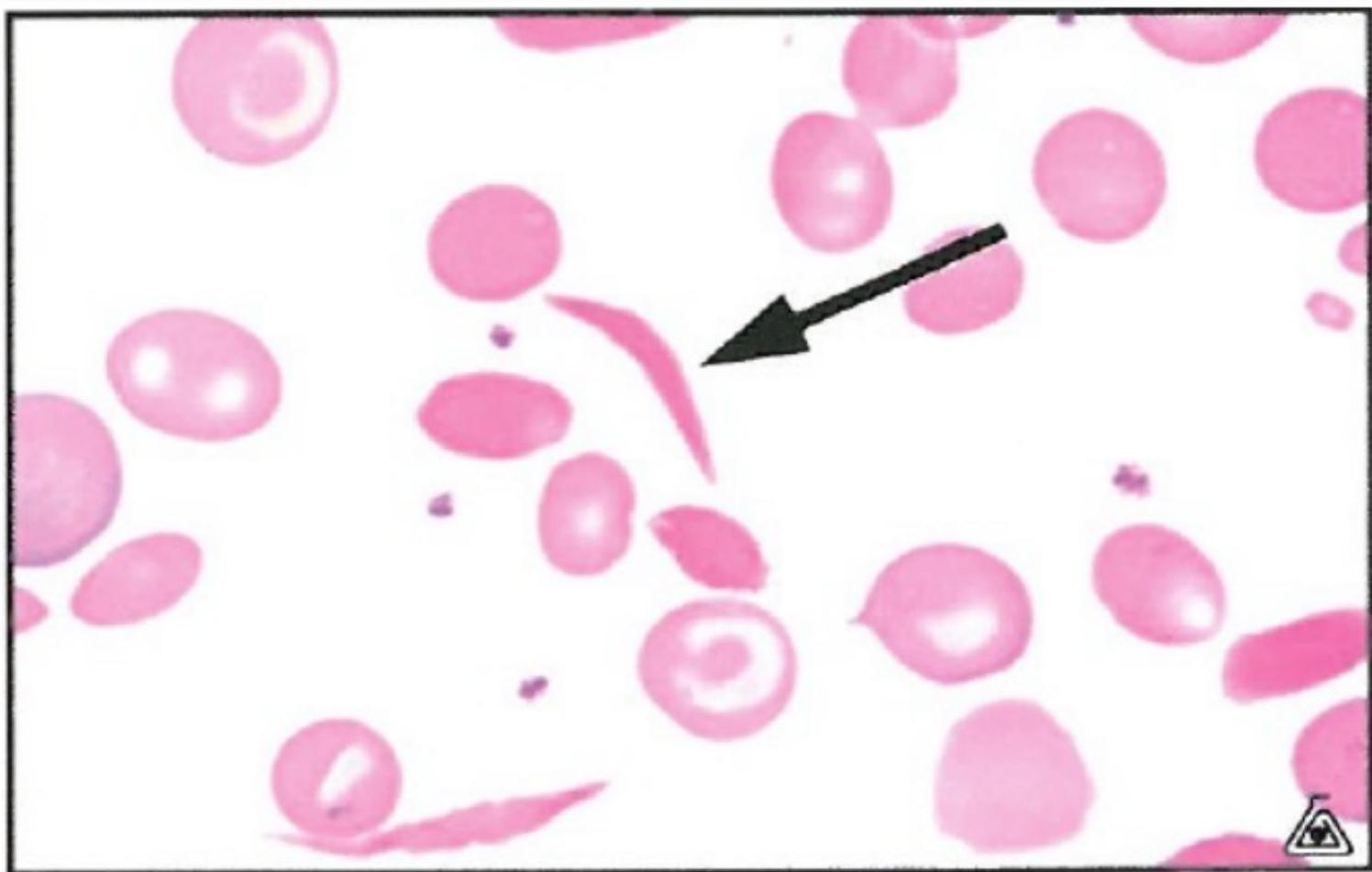
Schistocytes (aka fragments)
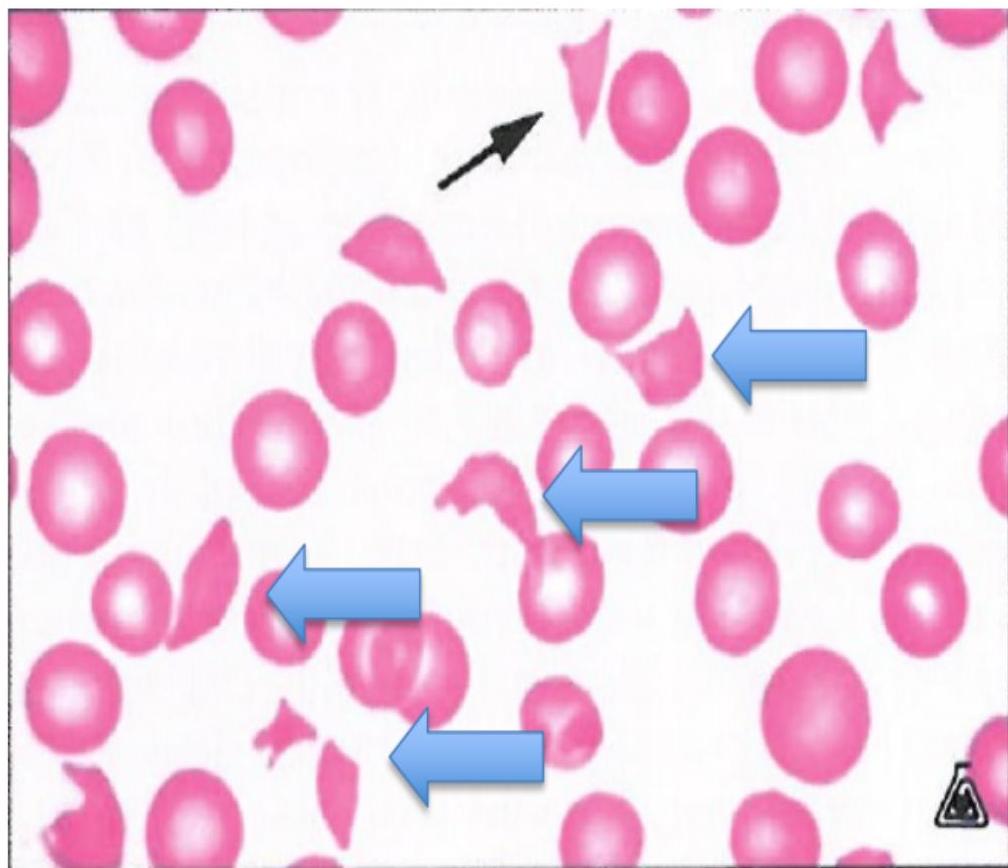
All arrows pointing to examples of schistocytes. Think TMA (eg. TTP)
Malaria
Sickle Cell
QUIZ 1
A 30-year-old woman is evaluated for decreased exercise tolerance and ice cravings for the past several weeks. Medical history is notable for Crohn colitis diagnosed 4 years ago. Her symptoms flared 3 months ago with increased abdominal pain and diarrhea, and she began therapy with azathioprine and infliximab. Her only other medication is ferrous sulfate tablets, 325 mg once daily, which she has been taking for 6 weeks after being diagnosed with iron deficiency anemia; her hemoglobin level at that time was 8.2 g/dL. On physical examination, vital signs are normal. She is thin, with pale conjunctivae and nail beds. Cardiac examination reveals a grade 2/6 systolic flow murmur. The remainder of the examination is unremarkable.
Laboratory studies show a hemoglobin level of 7.5 g/dL and a serum ferritin level of 1 ng/mL (12-150). Failure to respond to treatment
Which of the following is the most appropriate treatment?
A. Intravenous iron preparation B. Oral iron in a liquid preparation C. Oral iron tablets three times daily D. Sustained-release iron preparation
QUIZ 2
A 52-year-old man is evaluated in the emergency department for decreased exercise tolerance and yellowing of the eyes for the past 3 days. He was diagnosed with UTI 6 days ago and began antibacterial therapy 4 days ago with nalidixic acid. His complete blood count at that time was normal. On physical examination, temperature is 36.7 °C, blood pressure is 120/70 mm Hg, pulse rate is 100/min, and respiration rate is 18/min. He has icteric sclerae. Cardiac examination reveals a grade 2/6 systolic flow murmur. No organomegaly is observed.
Laboratory studies: Haptoglobin Undetectable Hemoglobin 5.6 g/dL Leukocyte count 5600/μL Platelet count 223,000/μL Reticulocyte count 12% of erythrocytes Examination of the peripheral blood smear shows bite cells. The direct antiglobulin (Coombs) test is negative.
Which of the following is the most appropriate immediate management?
A. Administer prednisone B. Administer folic acid C. Discontinue nalidixic acid D. Measure glucose-6-phosphate dehydrogenase level
QUIZ 3
A 56-year-old man is brought to the emergency department after being found lying unresponsive in the street. Medical history is significant for chronic alcohol dependence. His medical history is otherwise not significant. On physical examination, vital signs are normal. The patient is unkempt. He moans in response to painful stimuli and moves all extremities. He has poor dentition. Hepatomegaly is noted.
Laboratory studies:
- Hemoglobin 7.4 g/dL, Leukocyte count 4200/μL
- Mean corpuscular volume 110 fL, Platelet count 97,000/μL
- Reticulocyte count 1% of erythrocytes macocytic anemia
Hypersegmented neutrophils are seen on the peripheral blood smear.
Which of the following is the most likely cause of this patient’s anemia?
A. Cobalamin deficiency B. Folate deficiency C. Inflammatory anemia D. Iron deficiency
QUIZ 4
A 65-year-old woman is evaluated for slowly worsening fatigue and exertional dyspnea of several months’ duration, with more recent yellowing of the skin. Medical history is notable for Hashimoto thyroiditis diagnosed 35 years ago. Her only medication is levothyroxine.
On physical examination, temperature is 36.7 °C, blood pressure is 120/70 mm Hg, pulse rate is 100/min, and respiration rate is 18/min. Jaundice is noted. The remainder of the examination is noncontributory.
Laboratory studies:
- Hemoglobin 6.2 g/dL
- Leukocyte count 3100/μL, with a normal differential
- Mean corpuscular volume 110 fL
- Platelet count 94,000/μL
- Bilirubin: Total 6.2 mg/dL, Direct 1.0 mg/dL
- Lactate dehydrogenase 400 U/L (140-280)
A peripheral blood smear shows macrocytic erythrocytes with rare nucleated erythrocytes and scattered, six-lobed neutrophils. A direct antiglobulin (Coombs) test is negative.
Which of the following should be obtained next?
A. Bone marrow biopsy B. Cobalamin level C. Flow cytometry D. Glucose-6-phosphate dehydrogenase level
QUIZ 5
An 75-year-old woman is evaluated for fatigue and exertional dyspnea developing over several months. Medical history is significant for longstanding hypertension, but she has not been adherent with her medications.
On physical examination, vital signs are normal except for blood pressure of 170/110 mm Hg. She is frail, with pallor of mucous membranes and nail beds. The remainder of the examination is normal.
Laboratory studies: Hemoglobin 9 g/dL Leukocyte count 8700/μL, with a normal differential Mean corpuscular volume 85 fL Platelet count 380,000/μL Reticulocyte count 1% of erythrocytes Creatinine 1.8 mg/dL
CONT-
Folate 8 ng/mL (2.7-17) Iron studies: Ferritin 120 ng/mL (12-150) Iron 70 µg/dL (60-170) Total iron-binding capacity 250 µg/dL (240-450) Vitamin B12 540 pg/mL (160-950) Peripheral blood smear shows normal erythrocyte morphology. On kidney ultrasonography, kidneys are small bilaterally, with echographic features suggesting chronic kidney disease.
Which of the following is the most likely cause of this patient’s anemia? Anemia due to chronic kidney disease
A. Erythropoietin deficiency B. Inflammation C. Iron deficiency D. Myelodysplastic syndrome
QUIZ 6
A 20 year old lady with sickle cell disease (HbSS) presents with right limb weakness that progresses within hours to right sided hemiplegia. An urgent CT head confirms a new left ischemic stroke. She has a history of recurrent pain crises and had an episode of acute chest syndrome during an admission 5 years ago. Her hemoglobin is 9.7 g/dL, white blood cell count of 9000/μL and platelets of 490000/μL. She has been started on ASA and DVT prophylaxis, and morphine for pain control.
What should be done in addition to her current treatment?
a) 3L of normal saline b) Hydroxyurea c) Exchange transfusion d) 2 units RBCs
Thank You!