Inborn Errors of Metabolism
Overview
- Inborn errors of metabolism (IEMs) are rare genetic diseases due to a defect in an enzyme or transport protein which results in a block in a metabolic pathway leading to clinically significant consequences.
- Most are autosomal recessive.
- Incidence of IEM in S.A: 169 in 100,000 births (1:600), approximately 1000 cases per year.
- The incidence of all 16 disorders screened by the NBS Program in Saudi Arabia is nearly 1:1043 (highest incidences reported so far worldwide, 2017).
Basic Concept of Metabolic Disorder
Disorders in which there is a block at some point in the normal metabolic pathway caused by a genetic defect of a specific enzyme.
Types of Inborn Errors of Metabolism
Disorders of Protein Metabolism:
- Amino acids: Phenylketonuria, Maple Syrup Urine Disease
- Organic acids: Methylmalonic Aciduria, Propionic acidemia
- Urea cycle: Citrullinemia, Argininosuccinic Aciduria
Disorders of Carbohydrate Metabolism:
- Galactosemia
- Fructose intolerance
- Glycogen storage disease
Disorders of Fatty Acid Metabolism:
- Medium Chain Acyl CoA Dehydrogenase Deficiency (MCAD)
- Very Long Chain Acyl CoA Dehydrogenase Deficiency (VLCAD)
Lysosomal Storage Disorders:
- Gaucher’s disease
- Niemann–Pick disease
- Pompe disease
Disorders of Porphyrin Metabolism:
- Acute intermittent porphyria
Disorders of Purine or Pyrimidine Metabolism:
- Lesch–Nyhan syndrome
Disorders of Steroid Metabolism:
- Congenital adrenal hyperplasia
Disorders of Mitochondrial Function:
- Kearns–Sayre syndrome
Disorders of Peroxisomal Function:
- Zellweger syndrome
Peroxisomal Disorders
Zellweger Syndrome (Cerebro-hepato-renal Syndrome)
Clinical Signs
- Typical and easily recognized dysmorphic facies
- Progressive degeneration of Brain/Liver/Kidney, with death ~6 months after onset
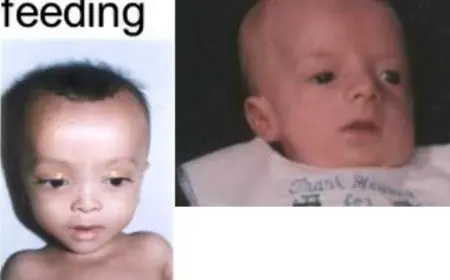
- Hypotonia, seizures and poor feeding
- Distinctive facies
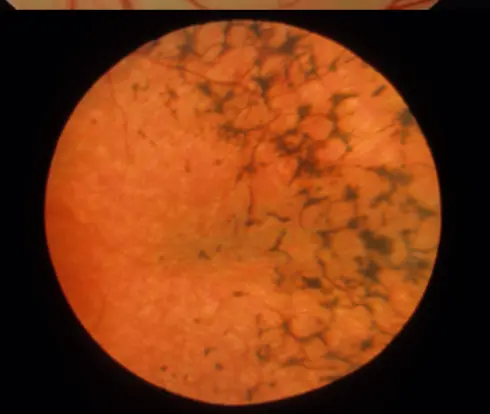
- Retinal dystrophy
- Hearing loss, severe developmental delay
Diagnosis
- Biochemical: serum Very Long Chain Fatty Acids (VLCFAs)
- Gene test
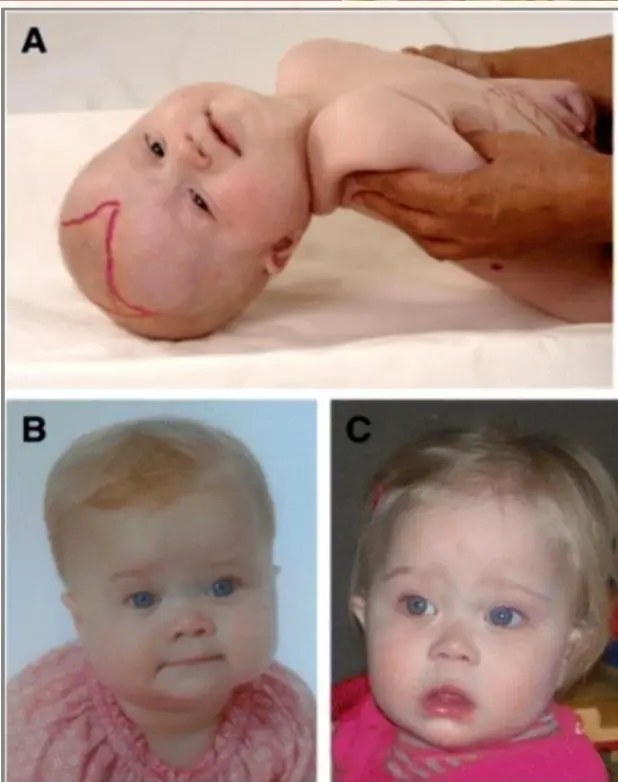
Zellweger Syndrome: Craniofacial Dysmorphism
- Separated cranial sutures
- Large anterior fontanelle
- High forehead
- Flattened face
- Low and broad nasal bridge
- Upslanting palpebral fissures
- Epicanthal folds, wide-set eyes
- Underdeveloped eyebrow ridges
- Protruding tongue
- Deformed ear lobes
- High arched palate
Age for Presentation of IEM
-
Presentation is usually in the neonatal period or infancy.
-
Age for presentation of clinical symptoms varies:
- Disorders of protein/carbohydrate metabolism: present in the neonatal period or early infancy
- Fatty acid oxidation defects and lysosomal storage disorders: present in infancy or childhood
-
Some IEM may be manifested by subtle neurologic or psychiatric features and often undiagnosed until adulthood.
-
The onset and severity may be exacerbated by environmental factors such as diet and intercurrent illness.
Clinical Manifestation
Acute: As Metabolic Emergencies
- Poor feeding, recurrent vomiting and dehydration
- Rapid deep breathing and apnea
- Lethargy, coma and seizures
- Sudden infant death syndrome (SIDS)
- Apparent life-threatening event (ALTE)
Chronic
- Dysmorphic features, failure to thrive
- Hepatomegaly, jaundice
- Cardiomyopathy (hypertrophic or dilated cardiomyopathy)
- Developmental delay or regression, spastic diplegia, ataxia, peripheral neuropathy
Ophthalmologic Manifestations
- Cataracts, corneal opacities or clouding
- Dislocated lenses
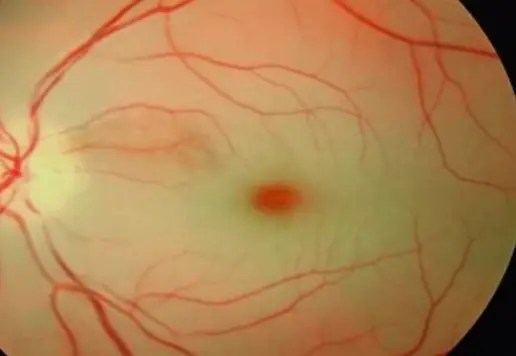
- Cherry-red spots
- Retinitis pigmentosa
Dermatologic Manifestations
- Rashes
- Photosensitivity
- Hyperkeratosis and ichthyosis
- Hypopigmentation, melanocytic nevi
Abnormal Odors
Abnormal odors in breath, urine, perspiration, saliva:
- Burnt sugar, curry, maple syrup – Maple syrup urine disease
- Sweaty socks or cheese-like – Isovaleric acidemia
- Fruity, ammoniacal – Methylmalonic or propionic acidemia
- Mouse urine, musty – Phenylketonuria
- Cabbage-like, rotten eggs – Tyrosinemia
- Malt or hops – Methionine malabsorption
- Cat urine – 3-methylcrotonic acidemia, 3-hydroxy-3-methylglutaric aciduria
- Fish-like – Trimethylaminuria and carnitine excess
Clinical Findings According to Age
Neonate
- Non-immune hydrops fetalis – e.g., Gaucher disease, MPS
- In neonates, findings may be indistinguishable from sepsis:
- Poor feeding, vomiting, diarrhea, and/or dehydration
- Temperature instability – hypo or hyperthermia
- Hypoglycemia
- Tachypnea, apnea
- Bradycardia
- CNS: Irritability, abnormal posturing, abnormal tone, seizures, and altered level of consciousness and coma
Infants and Young Children
- Dysmorphic or coarse features
- Recurrent vomiting, poor feeding
- Failure to thrive, developmental delay or regression
- Seizures, ataxia, lethargy, coma
- Skeletal abnormalities, abnormalities of hair or skin
- Dilated or hypertrophic cardiomyopathy
- Hepatomegaly, jaundice and liver dysfunction
- Visual and auditory disturbances
Older Children, Adolescents, and Adults
- Mild to profound mental retardation
- Autism, learning disorders, behavioral disturbances
- Hallucinations, delirium, aggressiveness, agitation, anxiety
- Panic attacks, seizures, dizziness, ataxia
- Exercise intolerance, muscle weakness, and paraparesis
Pompe Disease
- Glycogen storage disease type II (GSD II)
- Acid alpha-glucosidase (GAA) deficiency active in lysosomes
- Also known as acid maltase deficiency (AMD)
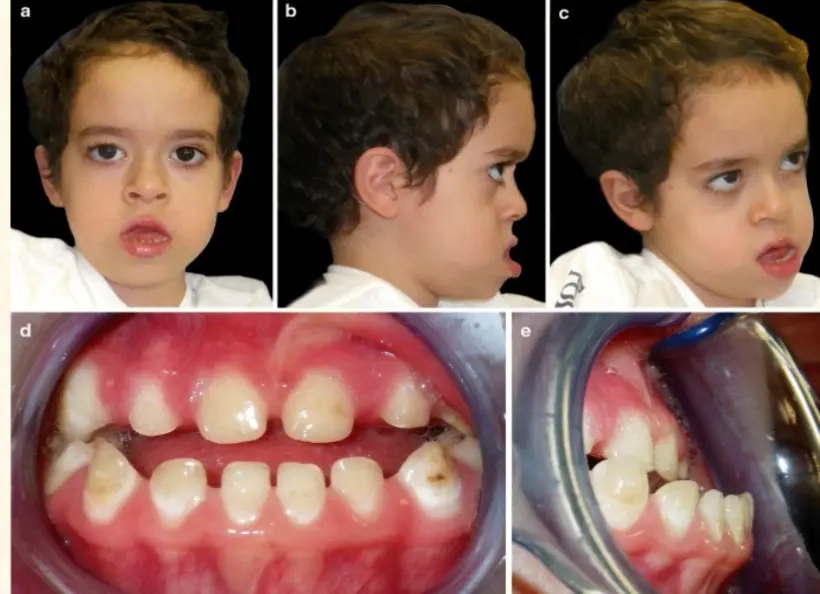
Clinical Features: Macroglossia, facial muscle weakness (myopathic facies), open mouth posture, drooling and dental malocclusions
Clinical Features by Onset
Infantile-onset: First Few Months of Life (~4 months)
- It is a rare, progressive, fatal, autosomal recessive lysosomal storage disorder
- Prevalence:
- Saudi Arabia: 1:5,000 – 1:14,000
- Global: 1 in 40,000
- Accumulation of glycogen within muscle cells, affecting the respiratory, musculoskeletal, cardiac, and GIT systems
- Cardiomyopathy and severe generalized muscular hypotonia (respiratory distress, muscle weakness, feeding difficulties and failure to thrive)
- Enlarged tongue, hepatomegaly and hypotonia
Late-onset: 2.6 to 81 years
- Delayed gross-motor development and progressive weakness in a limb-girdle distribution
- Do not develop cardiomyopathy
Signs and Symptoms
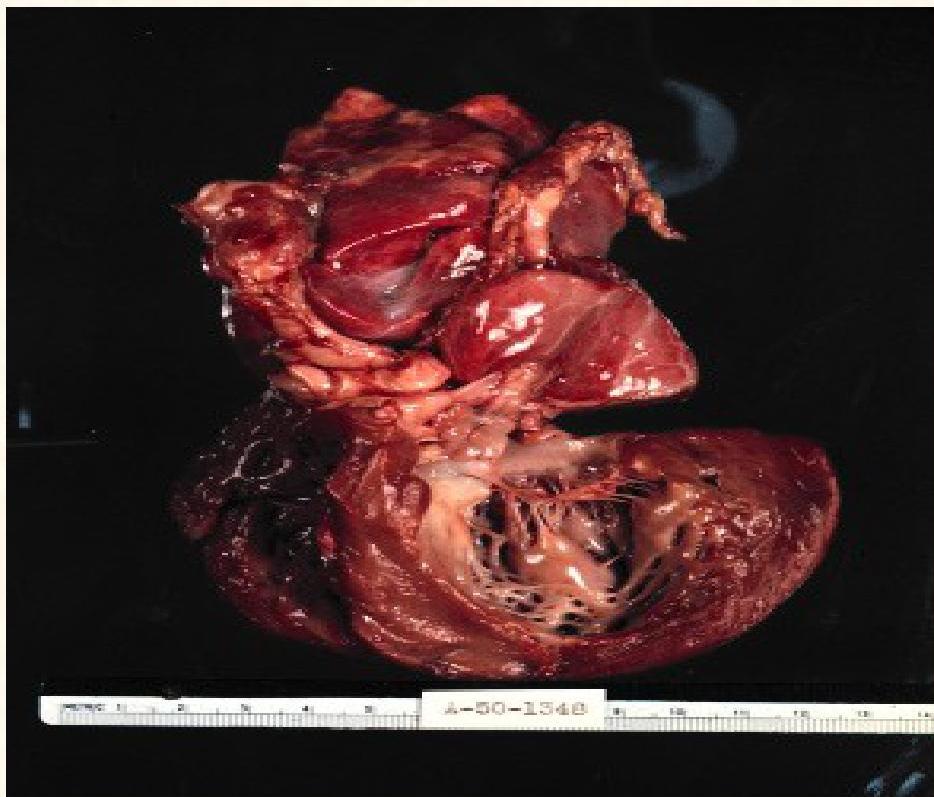
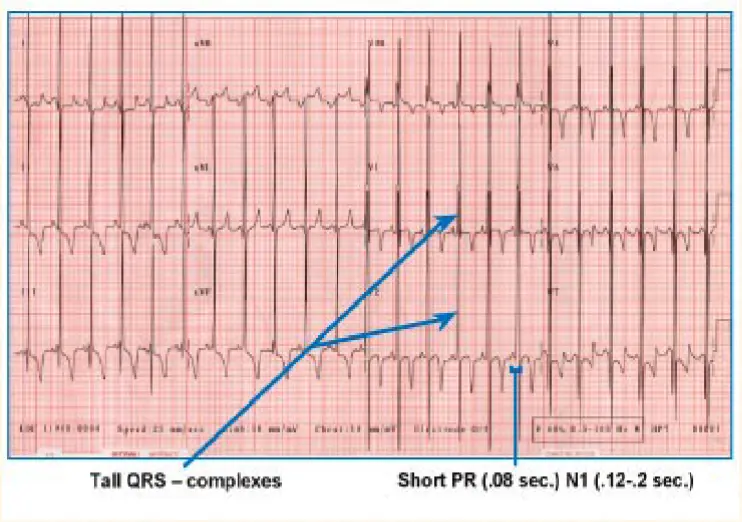
ECG Findings:
- Tall QRS-complexes
- Short PR (0.08 sec.) – Normal: (0.12-0.2 sec.)
Signs and Symptoms Summary
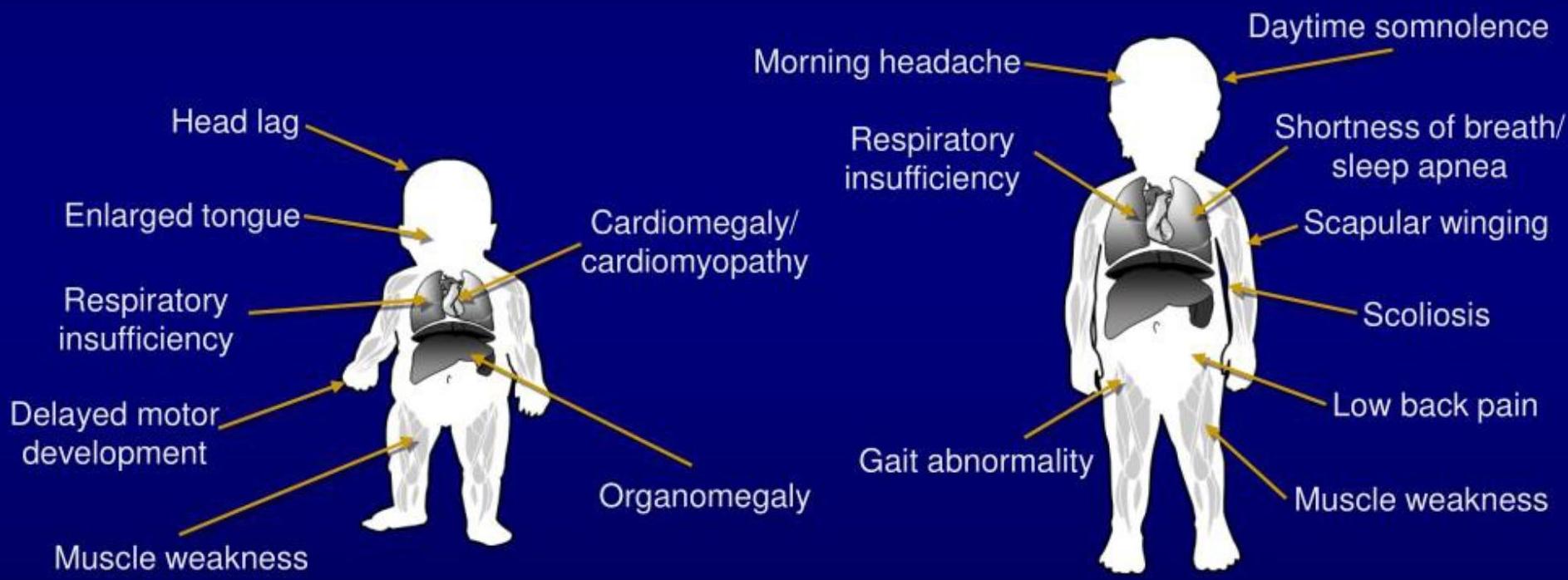
- Infantile onset: < 12 months
- Late onset: > 12 months
- Look for unusual symptoms or clusters of more common symptoms
Treatment
- Currently, the standard of care is enzyme replacement therapy (ERT)
- ERT has been proven to increase survival in Pompe disease as well as improve pulmonary and motor tests
- Gene therapy: In phase I/II trials
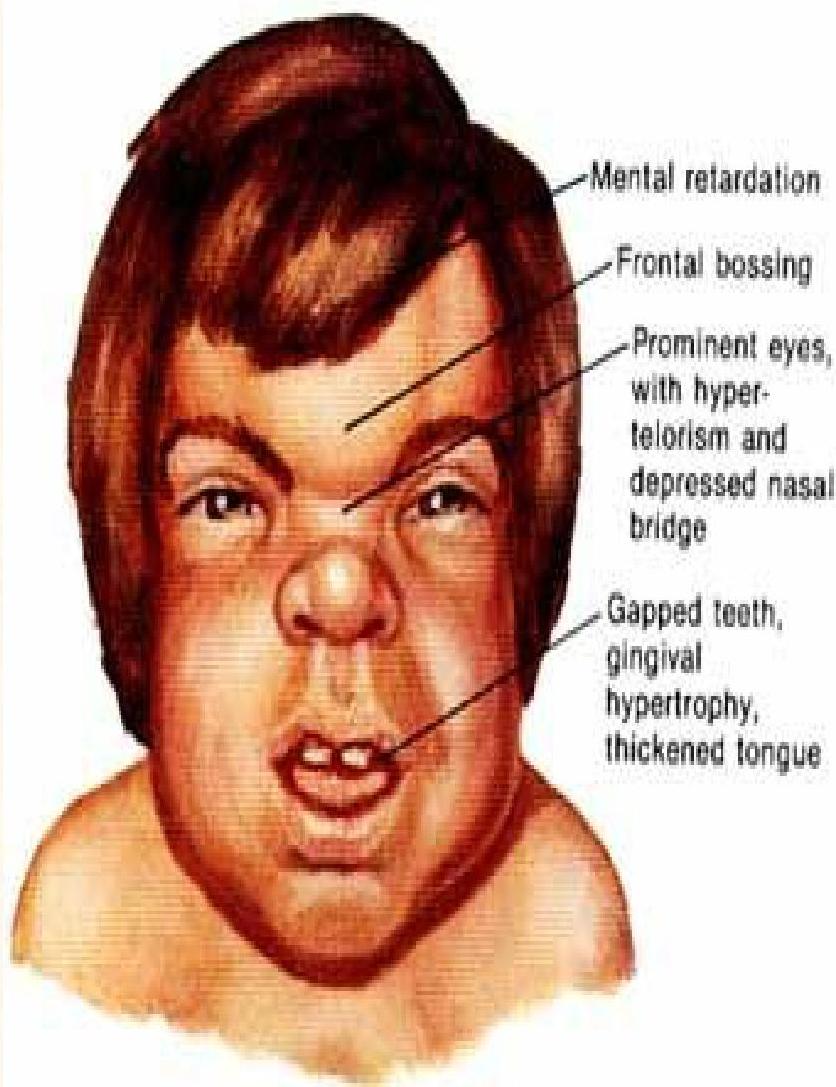
Mucopolysaccharidosis (MPS) -
Hurler Syndrome (Type 1 Mucopolysaccharidosis) Z OSPE
- A rare lysosomal storage disease
- Skeletal abnormalities, cognitive impairment, heart disease, respiratory problems, enlarged liver and spleen
- Characteristic facies
- Absence of alpha-L-iduronidase enzyme, responsible for degradation of MPS
- Build up of dermatan sulfate and heparin sulfate in multiple tissues, resulting in progressive deterioration and, eventually, death
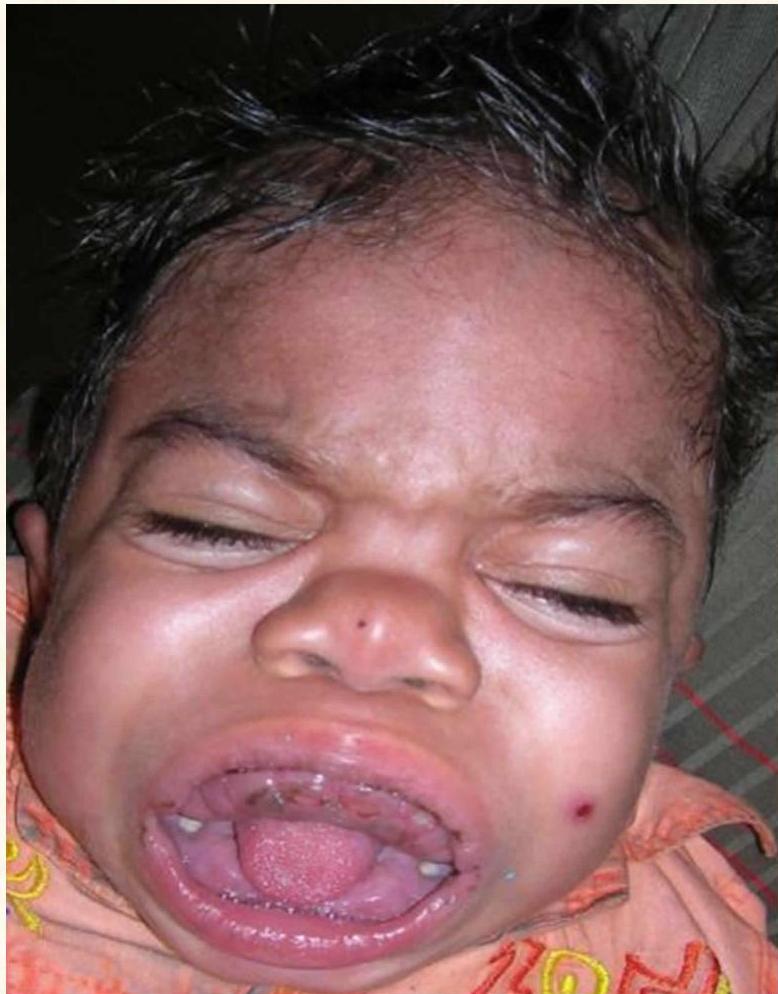
Hunter Syndrome (Type II Mucopolysaccharidosis)
- Lysosomal storage disease
- X-linked recessive
- Caused due to iduronate-2-sulfatase (I2S) enzyme deficiency, needed to break down glycosaminoglycans
- Facial changes, physical changes and intellectual disability seen by the age of 2-4 years old
{kind=link}
{kind=link}
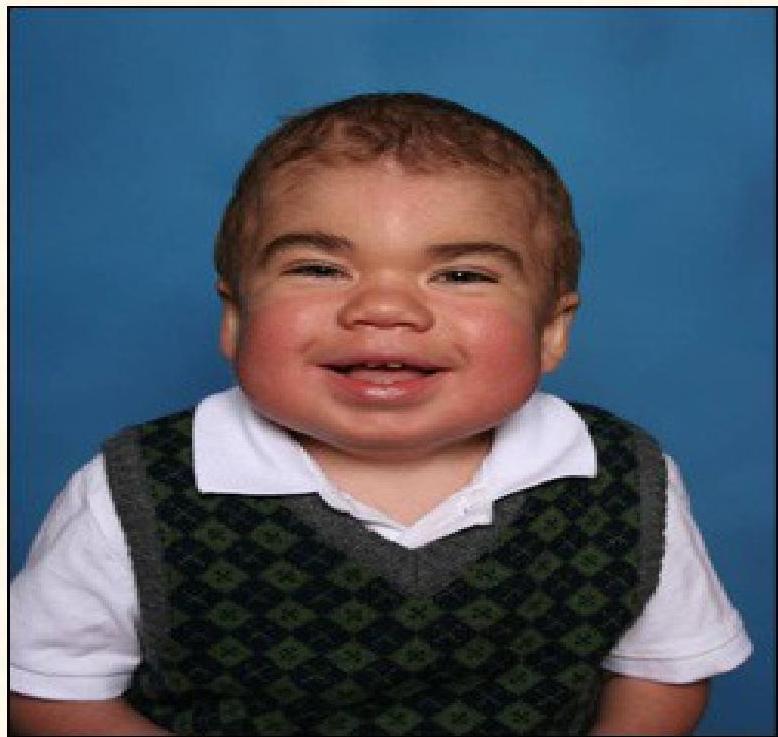
Hunter Syndrome Clinical Features
- Enlarged head, nose and tongue
- Round and hard cheeks, thick lips
- Hepatosplenomegaly, umbilical hernia
- Rigid joints
- Recurrent RTI (respiratory tract infections)
- Hydrocephalus
- Valve abnormalities
- Enzyme replacement therapy available
Diagnosis
Approach to Diagnosis
- History
- Physical examination
- Investigations
History
Any child with unexplained following signs and symptoms, IEM should be ruled out:
- Rapid deterioration in an otherwise well infant
- Septic appearing infant
- Persistent metabolic acidosis
- Hypoglycemia
- Unexplained mental retardation
- Developmental delay or regression, hypotonia
- Neurological deterioration, convulsion and coma
- Cardiomyopathy
- Hepatocellular dysfunction, hepatomegaly
- Failure to thrive
Family History
- Family history of consanguinity, ethnicity and inbreeding
- History of neonatal deaths or fetal losses in the family
- Maternal family history – e.g., X-linked disorders, mitochondrial disorders
- A positive family history may be helpful but a negative family history does not exclude the condition
Physical Examination
- The physical examination findings are nonspecific and may be normal
- Findings also vary with age and severity
- Abnormalities in general may include:
- Dysmorphic features
- Abnormalities of hair, skin, skeleton
- Abnormal odor
- Organomegaly
- Abnormal muscle tone
- Failure to thrive
Investigations
- Specimens of blood and urine for definitive diagnosis should be collected while the child is acutely ill
- Complete blood count
- Blood glucose, serum electrolytes and uric acid
- Blood urea nitrogen and creatinine
- Liver function: Serum bilirubin, transaminases, PT, aPTT, INR
- Blood gas and anion gap
- Plasma ammonia levels
- Urine: pH, ketones, and reducing substances levels and odor
- Lactate dehydrogenase, aldolase, creatinine kinase
- Urine myoglobin levels in patients with evidence of neuromyopathy
Newborn Screening: Heel Prick Test (Guthrie Test)
- Congenital Hypothyroidism (CHT)
- Sickle Cell disorders
- Cystic Fibrosis (CF)
- Inherited metabolic diseases (IMDs):
- Galactosemia
- Phenylketonuria (PKU)
- Medium-chain acyl-CoA dehydrogenase deficiency (MCAD)
- Maple syrup urine disease (MSUD)
- Isovaleric acidaemia (IVA)
- Glutaric aciduria type 1 (GA1)
- Homocystinuria (pyridoxine unresponsive) (HCU)
IEM and Endocrine Disorders Screened in Saudi Arabia National Newborn Screening Program

Aminoacidopathies
- Phenylketonuria (PKU)
- Argininosuccinatelyase deficiency (ASL)
- Maple syrup urine disease (MSUD)
- Citrullinaemia
Organic Acid Disorders
- Propionic acidaemia (PPA)
- Methylmalonicacidaemia
- Glutaricacidaemia type-I
- Isovalericacidaemia
- 3-methylcrotonyl-CoA carboxylase deficiency (3-MCC)
Fatty Acid Oxidation Defect
- Medium-chain acyl CoA dehydrogenase deficiency
Ketogenesis and Ketolysis Defects
- 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
Carbohydrate Disorder
- Beta-ketothiolase deficiency (BKT)
Endocrine Disorders
- Galactosaemia (GALT)
- Congenital hypothyroidism (CH)
- Congenital adrenal hyperplasia (CAH)
Tandem Mass Spectrometry Screening
- Tandem mass spectrometry allows screening for > 30 disorders, generally including:
- Aminoacidemias
- Urea cycle disorders
- Organic acidurias
- Fatty acid oxidation disorders
Specialized Tests
- Quantitative plasma amino acids: used to confirm the diagnosis of urea cycle disorders and other disorders of amino acid metabolism
- Qualitative urine organic acids by gas chromatography/mass spectrometry (GC/MS) – e.g., Organic acidemia
- Serum lactate and pyruvate: mitochondrial disorders, glycogen storage diseases, disorders of gluconeogenesis, and disorders of pyruvate metabolism
- Acylcarnitine profile: used for the diagnosis of fatty acid oxidation disorders
Specific Diagnostic Tests
- Tissue biopsy or autopsy: liver, muscle, brain, bone marrow
- Skin biopsy and fibroblast cultivation: for specific enzyme testing
- Molecular genetic testing: Specific DNA testing is available for certain IEM
Differential Diagnosis
In Neonates
- Sepsis
- Congenital viral infection
- Duct-dependent congenital heart disease
- Drug withdrawal
- Congenital adrenal hyperplasia
In Older Children
- Diabetes
- Drug ingestion or intoxication
- Encephalitis
- Adrenal insufficiency
Management Approach
Goals of Treatment
- Prevention of further accumulation of harmful substances
- Management of complications and correction of metabolic abnormalities
- Elimination of toxic metabolites
- Treatment should be initiated as quickly as possible because apparently stable patient with mild symptoms may deteriorate rapidly, with progression to death
- With appropriate therapy, patients may completely recover without any complication
General Management
In any critically ill child, appropriate and aggressive treatment before confirmation of the diagnosis may be life-saving:
- Assessment of airway, breathing and circulation (ABC)
- If shock: IV normal saline bolus (Lactated ringers solution should be avoided)
- Correct hypoglycemia, prevent catabolism
- Promote urinary excretion of toxic metabolites (1.5 times maintenance fluid)
- Correct acidosis, electrolyte abnormalities
- Correct hyperammonemia (sodium phenylacetate, sodium benzoate and hemodialysis)
- Consider broad spectrum antibiotics
- Discontinue oral intake in patients with decreased level of consciousness or vomiting
- Eliminate intake or administration of potentially harmful protein or sugars – e.g., galactose and fructose
- In case of known IEM, disease-specific agents should be eliminated
- Provide cofactors – e.g., cobalamin, biotin, carnitine and riboflavin
- In patient with seizure: Diazepam or phenobarbitone, Pyridoxine (B6)
Follow-up Management
- Once toxic metabolites have been normalized, protein can be reintroduced using an essential amino acid solution, initially at 0.5-0.75 g/kg/day and gradually increased
- For amino and organic acidopathies and urea cycle defects, protein intake should be restricted to 40-50% of recommended daily allowance
- Lipids, 2-3 g/kg/day as 20% intralipid, can be given to increase caloric intake, but they are contraindicated for certain fatty acid oxidation defects
- Pharmacologic therapy may be needed to increase activity of abnormal cofactor-dependent enzymes – e.g., thiamine [B-1], biotin, riboflavin [B-2], cobalamin [B-12]
- Diet for IEM include medical foods and dietary supplements along with dietary modifications excluding the IEM specific nutrients
Specific Management
- Bone marrow transplantation
- Organ transplantation
- Enzyme replacement therapy
- Gene therapy
Phenylketonuria (PKU) z
Overview
- Deficiency of phenylalanine hydroxylase z (enzyme for conversion of phenylalanine to tyrosine)
- Autosomal recessive inheritance
- Prenatal diagnosis: by amniocentesis or chorionic villus sampling, with identification of the gene
- Screening: Guthrie Test, tandem mass spectrometry
- Measurement of phenylalanine: High Phenylalanine > 20 mg/dl; High phenyl pyruvic acid
Symptoms
- Mental retardation
- Seizures
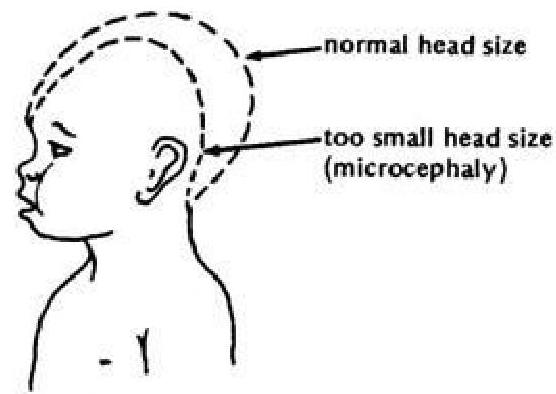
- Microcephaly (small head size) z
- Skin rashes
- Stunted growth
- Hyperactivity
- “Musty” body odor from the excess phenylalanine
- Fair skin, hair, and eyes (phenylalanine is linked to melanin production)
Clinical Features
- At birth, the neonate appears normal and asymptomatic prior to the initiation of feeds
- Blond hair, blue eyes, fair skin, and photosensitivity
- Seborrheic dermatitis or eczema skin
- Unpleasant musty odor of phenyl acetic acid
- Hyperactivity, athetosis, vomiting
- Seizures, hypertonia
- Severe mental retardation
Treatment
- The mainstay of therapy is dietary restriction of phenylalanine for life z
- Treatment should be started as soon as possible, usually before one week of age
- Early supplementation of low phenylalanine diet – the skin color, photosensitivity, odor, and eczema are reversible
- Low phenylalanine diet started early in infancy can dramatically reduce the mental retardation
- Supplementation with tyrosine or tryptophan in the diet may be necessary
- BH4 (Tetrahydrobiopterin)
- L-dopa and 5-hydroxytryptophan
Treatment Targets
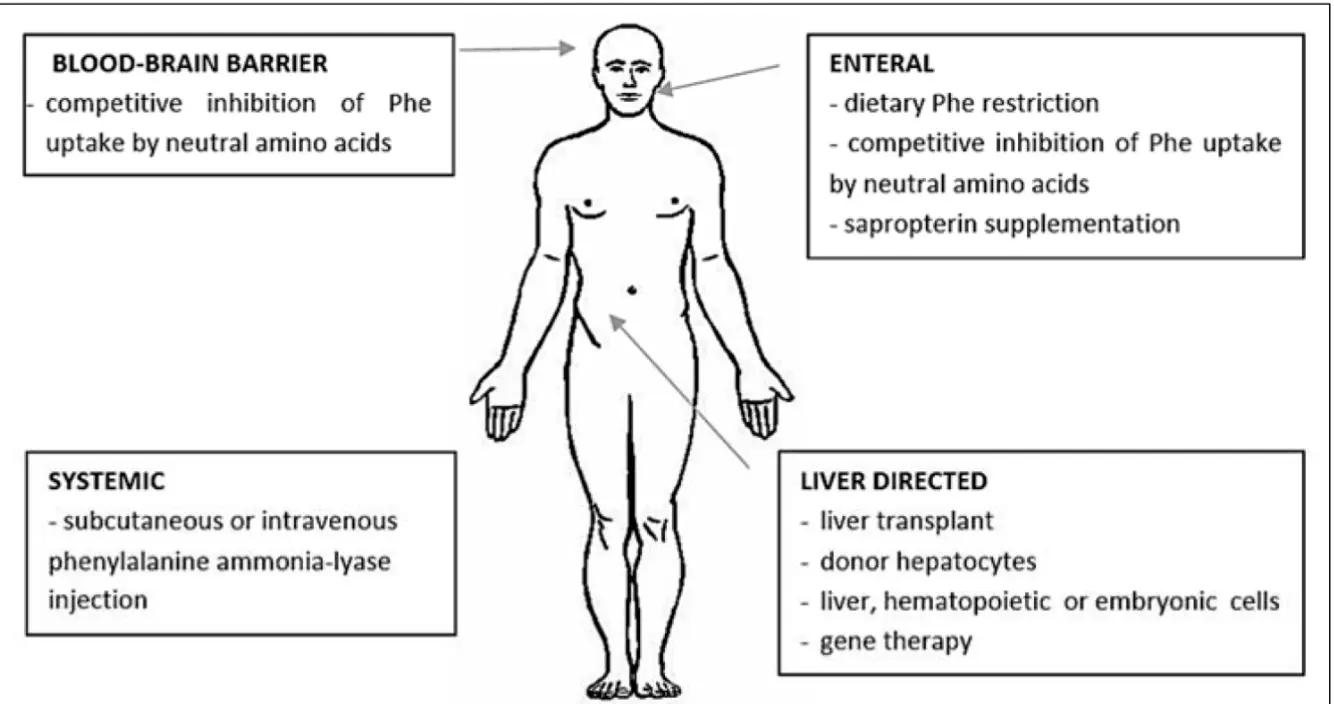
Blood-Brain Barrier
- Competitive inhibition of Phe uptake by neutral amino acids
Enteral
- Dietary Phe restriction
- Competitive inhibition of Phe uptake by neutral amino acids
- Sapropterin supplementation
Systemic
- Subcutaneous or intravenous phenylalanine ammonia-lyase injection
Liver Directed
- Liver transplant
- Donor hepatocytes
- Liver, hematopoietic or embryonic cells
- Gene therapy
Prognosis
- If untreated, PKU results in mental retardation, seizures, psychosis, hyperreflexia, and growth retardation
- Dietary treatment appears to reverse all signs of PKU except cognitive impairment that has already occurred
- Treated PKU patients typically have a lower IQ than the general population
- For women with PKU, dietary restriction must be started before conception and continued throughout pregnancy
Methylmalonic Acidemia (MMA) Z
Overview
- It is a disorder of amino acid metabolism, classified as organic acidemias
- Autosomal recessive inheritance
- Elevation of methylmalonic acid in the blood and/or the urine due to defect in metabolism of methylmalonyl-coenzyme A (CoA) or cobalamin
- Due to deficiencies in methylmalonyl-CoA mutase activity and in enzymatic synthesis of cobalamin, which lead to defect in the conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA
Clinical Features
- Children may be healthy at birth and develop symptoms soon after starting protein intake
- Typical presentation at the age of 1 month to 1 year may include:
- Vomiting, dehydration, lethargy, seizures, recurrent infections, and progressive encephalopathy
- On examination, child may have hypotonia, failure to thrive, hepatosplenomegaly
Investigations
- Metabolic ketoacidosis is the clinical hallmark
- Sample of blood and urine should be collected and sent for metabolic screening by Gas chromatography-mass spectrometry
- Blood levels of ammonia, glycine, and methylmalonic acid are elevated
- Definitive diagnosis is made after specific enzyme analysis of fibroblasts
Treatment
- Halt catabolism and the production of toxic organic acids by providing high-calorie intravenous dextrose (and often insulin to promote anabolism), while eliminating the protein/precursor source
- Correct the acidosis and hyperammonemia in acute phase
- Treat the intercurrent infection
- Long term therapy consists of protein restriction and pharmacologic doses of vitamin B12 and betaine
Galactosemia Z
Overview
- Altered metabolism of galactose due to deficient activity of one of 3 enzymes results in elevated blood galactose concentration (galactosemia)
- Autosomal recessive inheritance
- Incidence: 1 in 60,000 live births
- Galactose is a sugar found in human and bovine milk, as part of the disaccharide lactose. Lactose is hydrolyzed to glucose and galactose by the intestinal enzyme lactase.
- Classic galactosemia: complete deficiency of galactose-1-phosphate uridyl transferase (GALT), most common and severe type
Clinical Manifestations
Early Manifestations
- Vomiting, diarrhea
- Hepatomegaly and jaundice
- Failure to thrive, poor feeding, lethargy
- Sepsis (Escherichia coli)
- Cataracts may be present at birth but generally appear after two weeks
Late Manifestations
- Mental retardation (common), speech and language problems
- Ovarian failure
- Liver cirrhosis
- Severe failure to thrive
Laboratory Findings
- Increased plasma galactose and RBC galactose-1-P concentration, increased blood and urine galactitol levels
- Hypoglycemia due to lethargy, poor feeding, and liver dysfunction
- Liver dysfunction: Conjugated and unconjugated hyperbilirubinemia, elevated transaminases, coagulopathy, increased levels of plasma amino acids (especially phenylalanine, tyrosine, and methionine)
- Renal tubular dysfunction: Metabolic acidosis, galactosuria (reducing substances in the urine), glycosuria, aminoaciduria, albuminuria
- Hemolytic anemia
Investigations
- Newborn screening: fluorometric assay of galactose-1-phosphate uridyl transferase (GALT) enzyme activity in red blood cells
Diagnosis
- Assisted by reducing substances in urine
- Confirmation by Galactose-1-PO uridyl transferase activity in RBCs
- Prenatal diagnosis can be made by GALT assay in fibroblasts cultured from amniotic fluid or chorionic villus biopsy
- Genetic testing: GALT gene, located on chromosome 9p13
Treatment and Prognosis
- Lactose-free formula followed by dietary restriction of all lactose-containing foods in later life (tomatoes, brussels sprouts, bananas, and apples)
- Human milk or formula based on cow’s milk should be discontinued and a soy-based formula is to be given
- The early signs and symptoms, such as liver dysfunction, susceptibility to infections, failure to thrive, and cataracts, can be prevented or improved by early diagnosis and treatment, but patients can still have chronic and progressive neuropsychiatric impairments z
Fatty Acid Oxidation Defects
Overview
- Fatty acid oxidation defects are disorders of lipid metabolism
- Caused by a lack or deficiency of the enzymes needed to break down fats
Types of Fatty Acid Oxidation Defects:
- Very long-chain acyl-coenzyme A dehydrogenase (VLCAD)
- Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD)
- Medium-chain acyl-coenzyme A dehydrogenase (MCAD)
- Short-chain acyl-coenzyme A dehydrogenase (SCAD)
- Short-chain 3-hydroxyacyl-coenzyme A dehydrogenase (SCHAD)
Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCAD)
- Incidence: one in 5,000 - 10,000 live births (most common)
- Autosomal recessive
Clinical Features:
- Infants with MCAD deficiency appear to develop normally
- Present with rapidly progressive hypoketotic hypoglycemia, lethargy, seizures and coma, secondary to acute vomiting or fasting
- MCAD deficiency frequently manifests with sudden and unexpected death
- Diagnosed through newborn screening, by plasma acylcarnitine analysis and molecular genetic testing
Treatment of MCAD Deficiency
- The most important intervention is giving simple carbohydrates by mouth during acute manifestation
- Frequent cornstarch feeds and avoidance of fasting
- Relatively low-fat diet (e.g., less than 30% of total energy) in toddler
- The prognosis is excellent once the diagnosis is established
Maple Syrup Urine Disease (MSUD)
Overview
- Gene defect: People with this disease are missing the enzyme BCKD (branched chain-alpha ketoacid-dehydrogenase)
- Autosomal recessive disease
- Incidence: 1 in 180,000 births
- Maple Syrup Urine Disease is a genetic disease in which the amino acids leucine, isoleucine and valine cannot be broken down by branched-chain alpha-keto acid dehydrogenase
- Reference: http://www.msud-support.org/overv.htm
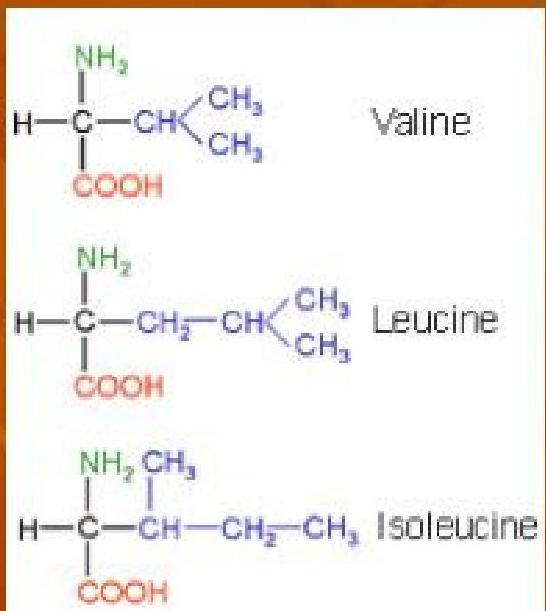
Amino Acid Structure: Amino acids contain an alpha carbon (C), an amino group (NH₃), a carboxyl group (COOH), and a unique side group (R). All branched-chain amino acids have side groups that contain a branched carbon chain.
Symptoms and Effects
- Poor feeding
- Vomiting
- Lack of energy
- Seizures
- Mental health problems
- Physical retardation
- Mental retardation
- Sweet odor, much like burned sugar

Screening and Diagnosis
- As with PKU, prenatal diagnosis and neonatal screening are available, and most affected individuals are compound heterozygotes
Treatment
-
The disease is treated with a synthetic formula that contains limited amounts of leucine, isoleucine, and valine—sufficient to provide the branched-chain amino acids necessary for normal growth and development without producing toxic levels
-
Early diagnosis and lifelong dietary treatment is essential if the child with MSUD is to develop normally
“وَفِي أَنْفُسِكُمْ أَفَلَا تَنْصُرُونَ”
(And in yourselves, then will you not see?)