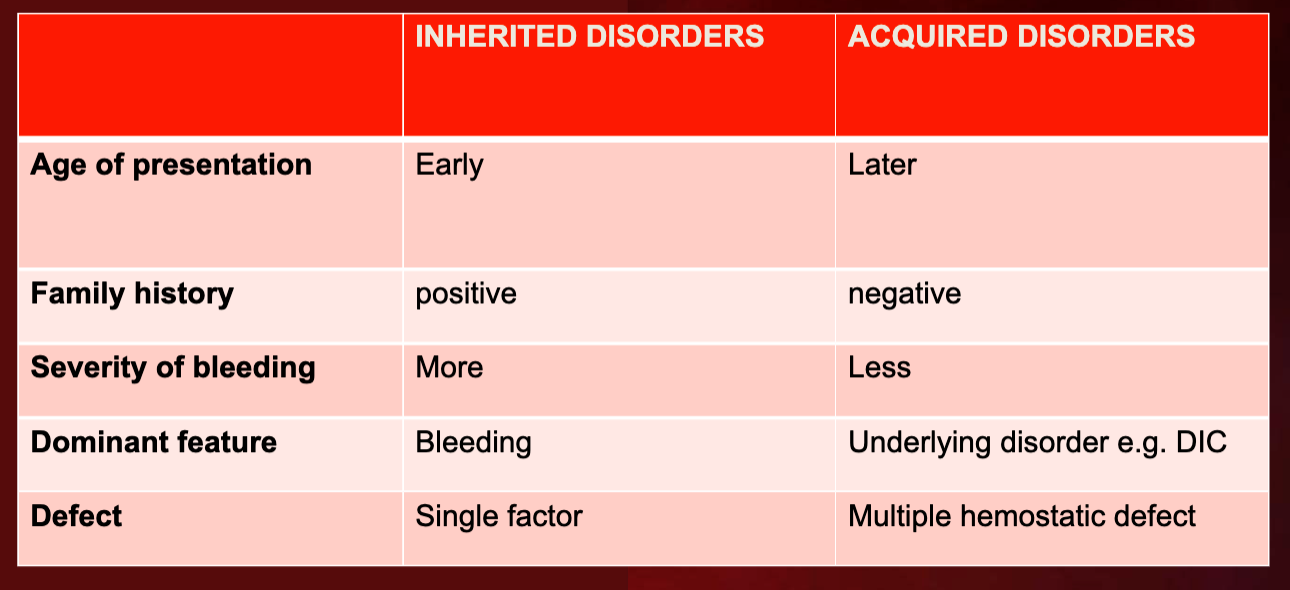
Hemophilia
Types of Hemophilia
-
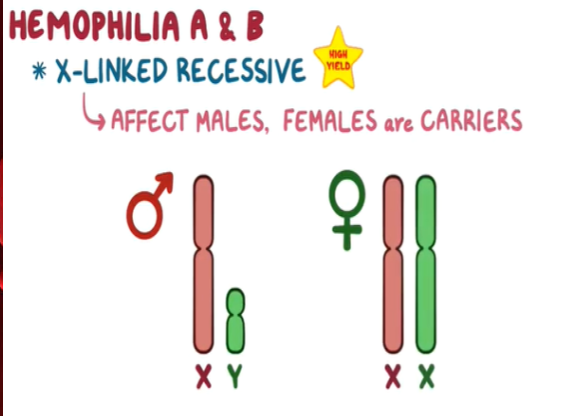
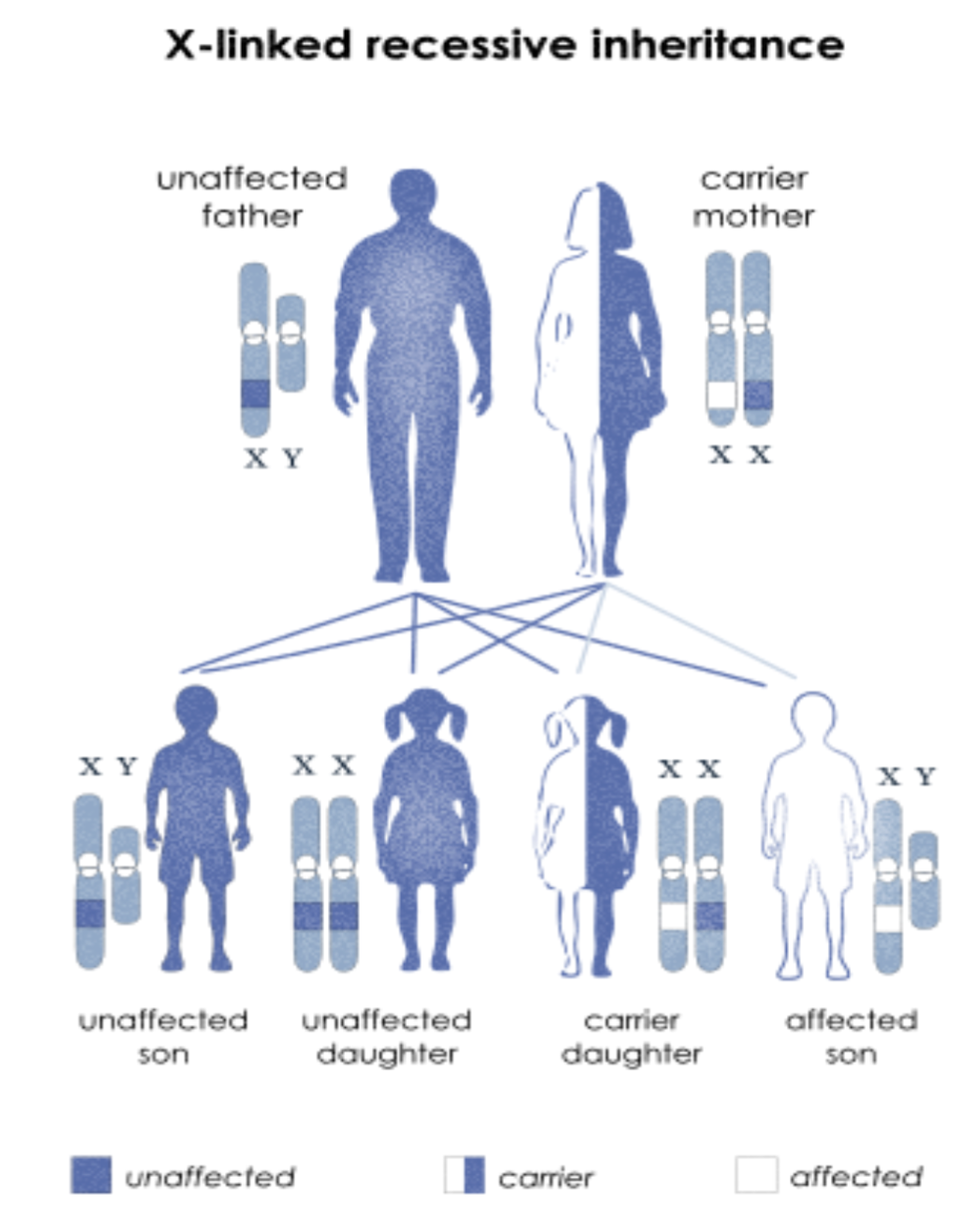
Hemophilia A & B: X-linked, recessive disorders caused by deficiency of functional plasma clotting factor VIII (A-8) (FVIII) or IX (9) (FIX), respectively. Can be inherited or acquired.
-
Hemophilia C: Autosomal recessive disorder, affecting both males and females, leading to a deficiency in factor XI(11).
-
Acquired Hemophilia A: Development of inhibitory antibodies to FVIII (8) complicates the treatment of genetic cases.
Hemophilia B
Hemophilia B, or Christmas disease, is an inherited, X-linked, recessive disorder that results in deficiency of functional plasma coagulation factor IX.
Hemophilia B accounts for 20% of hemophilia cases, and about 50% of those affected have factor IX levels greater than 1%.
Severity, Factor Activity, and Hemorrhage
| Hemophilia A | Hemophilia B | |
|---|---|---|
| Factor Deficiency | Factor VIII | Factor IX |
| Inheritance | X-linked recessive | X-linked recessive |
| Incidence | 1/10,000 males (80% of cases) | 1/50,000 males |
 |
Coagulation Disorder Symptoms
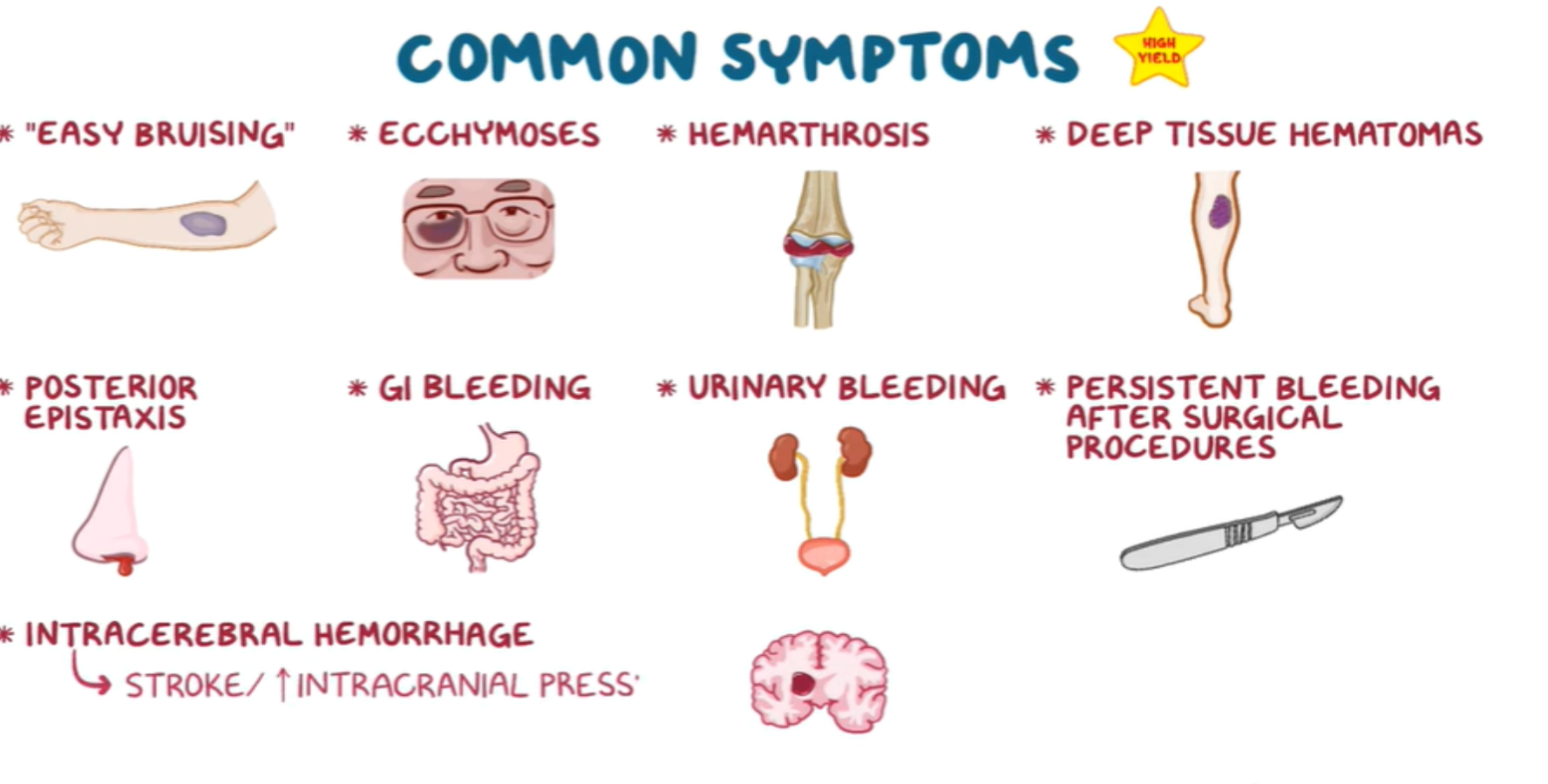
Common Symptoms:
- “Easy bruising”
- Ecchymoses
- Hemarthrosis
- Deep tissue hematomas
- Posterior epistaxis
- GI bleeding
- Urinary bleeding
- Persistent bleeding after surgical procedures
- Intracerebral hemorrhage (→ stroke / increased intracranial pressure)
Clinical Manifestations of Bleeding Disorders
| Bleeding Symptoms | Platelet Defects (qualitative or quantitative) | Clotting Factor Deficiencies (factor VIII or factor IX) |
|---|---|---|
| Overview of Bleeding Events | Mucocutaneous bleeding (oral, nasal, GIT, vaginal, and genitourinary sites) | Deep tissue bleeding (including joints and muscles) |
| Excessive Bleeding After Minor Cuts | Yes | Not usually |
| Epistaxis | anterior | posterior |
| Petechia | Common | Uncommon |
| Ecchymoses | small and superficial | large subcutaneous and soft tissue hematomas |
| Hemarthroses, Muscle Hematomas | Uncommon | Common (severe deficiency) |
| Bleeding with Invasive Procedures, Including Surgery | immediate, dependent upon the severity of defect, ranging from mild to severe | Either with procedural bleeding or delayed bleeding |
Hemophilia Diagnosis
Laboratory Studies
- CBC:
- Hemoglobin/hematocrit: Normal or decreased
- Platelet count: Normal
- Coagulation Studies:
- Bleeding time: Normal
- Prothrombin time (PT): Normal (Intrinsic)
- Activated partial thromboplastin time (aPTT): prolonged (mixed study)
- FVIII, IX, XI Assays
- Genetic Testing: Screening
Diagnostic Tests
- Prolonged aPTT (High Yield) (Rule out von Willebrand disease)
- Normal PT & platelet count (High Yield)
- Mixing Study
- aPTT prolongation due to true factor deficiency or presence of factor inhibitor
- If aPTT normalizes after mixing with normal plasma → factor deficiency (hemophilia)
- If aPTT does not normalize after mixing with normal plasma → factor inhibitor - Acquired

Hemophilia Management
Non-pharmacological Management
- Physiotherapy
- Hydrotherapy
- Patient education on treatment and prevention of bleeding
- Avoid non-steroidal anti-inflammatory drugs (NSAIDs) and intramuscular (IM) / joint injections
Pharmacological Management of different cases
-
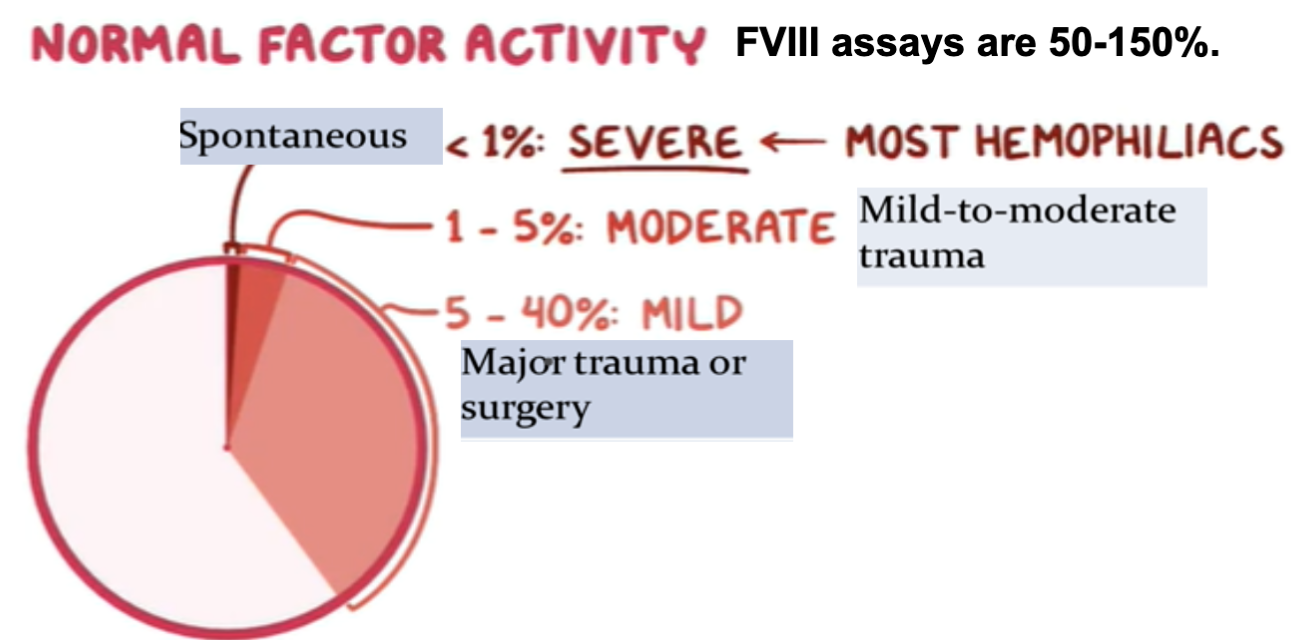
Mild Hemorrhages:
- Early hemarthrosis
- Epistaxis
- Gingival bleeding
- FVIII level of at 30%
-
Major Hemorrhages:
- Hemarthrosis
- Muscle bleeding
- Prophylaxis after head trauma with negative findings on examination
- FVIII level of at least 50%
-
Life-threatening Bleeding:
- Major trauma
- Surgery
- Advanced or recurrent hemarthrosis
- FVIII level of 80-100%
Medications
-
Desmopressin (DDAVP): Vasopressin analog used for mild hemophilia A. Not effective for severe hemophilia. Peak effect observed in 30-60 minutes. Intranasal spray available for outpatient use.
-
Emicizumab: Humanized monoclonal bispecific antibody that reduces the risk of bleeding events in hemophilia A. Bridges activated factor IX and factor X by binding to both factors (thereby replacing the deficient factor VIII), leading to activation of factor X and restoration of the clotting cascade.
-
Antifibrinolytics (e.g., tranexamic acid): Used in conjunction with FVIII replacement for oral mucosal hemorrhage and prophylaxis.
-
Factor Replacement Therapy: IV InfusionZ
- Hemophilia A: IV infusion of factor VIII concentrate.
- Hemophilia B: IV infusion of factor IX concentrate (has a longer half-life than factor VIII, so transfusions are less frequent).
- Hemophilia C: IV infusion of factor XI concentrate.
Note: Needed number of units of FVIII is calculated by the formula: (weight ÷ 4.4) × (factor level desired) = number of factor VIII units neededX
Inhibitors - Acquired hemophilia
Approximately 30% of people with hemophilia develop an antibody to the clotting factor they receive. These antibodies are known as inhibitors, often secondary to antiphospholipid syndrome (APS).
- Testing for Inhibitors: Bleeding is not controlled after adequate amounts of factor concentrate are infused during a bleeding episode.
- Treatment: High doses of FVIIa for bleeds or surgery. This overrides the defect in FVIII or FIX deficiency.
- Long-term Management: Eradicate inhibitors by administering high-dose FVIII (or FIX) in a process called immune tolerance.
Prognosis
- Prognosis is dependent on severity.
- Up to 60% of individuals with hemophilia A have severe forms.
- Prognosis is improved with proper treatment and prevention of injuries.
- Genetic therapies are currently under development and may further improve prognosis.
Complications
-
Complications of Disease:
- Degenerative joint disorders (arthritis)
- Life-threatening hemorrhage
-
Complications of Treatment:
- Viral infections (e.g., hepatitis, HIV) from transfusion
- Development of antibodies against the administered coagulation factors
- Opiate addiction