Internal Medicine
Dr. M. A. SOFI
What is Hemoglobin?
Hemoglobin is the iron-containing oxygen-transport metalloprotein in the red blood cells of all vertebrates. It plays a crucial role in delivering oxygen from the lungs to the body’s tissues.
Understanding Thalassemia
The normal hemoglobin molecule has a heme base surrounded by two pairs of globin chains. There are four types of globin chains:
- Alpha (α)
- Beta (β)
- Gamma (γ)
- Delta (δ)
Different combinations of these chains form different types of hemoglobin:
- HbA: The most common form of adult hemoglobin, with two α and two β chains.
- HbF: Fetal hemoglobin, with two α and two γ chains (predominant before birth).
- HbA2: Present in smaller amounts, with two α and two δ chains.
What is Thalassemia?
Thalassemias are a group of recessively autosomal inherited conditions characterized by decreased or absent synthesis of one of the two polypeptide chains (α or β) that form the normal adult human hemoglobin molecule (HbA, α2β2).
- β-globin gene defects: Give rise to β-thalassemia.
- α-globin gene defects: Cause α-thalassemia.
Over 300 mutations can cause thalassemia, leading to varying clinical severity. Thalassemia major, intermediate, and minor are terms used to describe the disease’s severity.
Thalassemia: Epidemiology
Approximately 5% of the worldwide population has a variation in the alpha or beta part of the hemoglobin molecule, though not all are symptomatic. Those without symptoms are called silent carriers.
- α-thalassemia: Prevalent in Southeast Asia, Africa, and India.
- β-thalassemia: Prevalent in areas around the Mediterranean, the Middle East, and Central, South, and Southeast Asia.
Increased migration has led to a rise in thalassemia gene mutations worldwide.
Types of thalassemia
Investigations
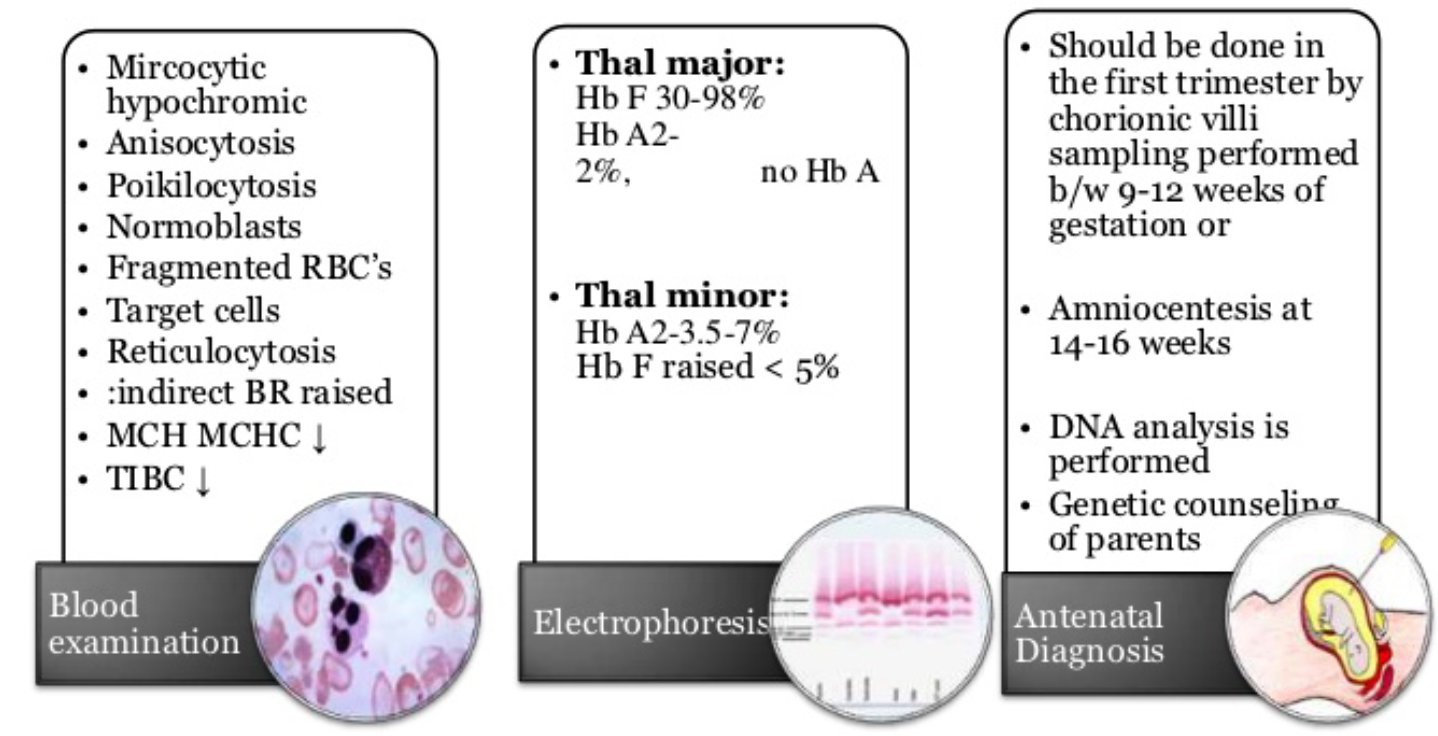
Blood Tests
- FBC: Shows microcytic, hypochromic anemia.
- Hemoglobin level: Ranges from 2-8 g/dL in severe forms.
- WBC: Elevated due to hemolytic process.
- Platelet count: May be depressed in splenomegaly.
- Serum iron level: Elevated with saturation as high as 80%.
- Ferritin: Also raised.
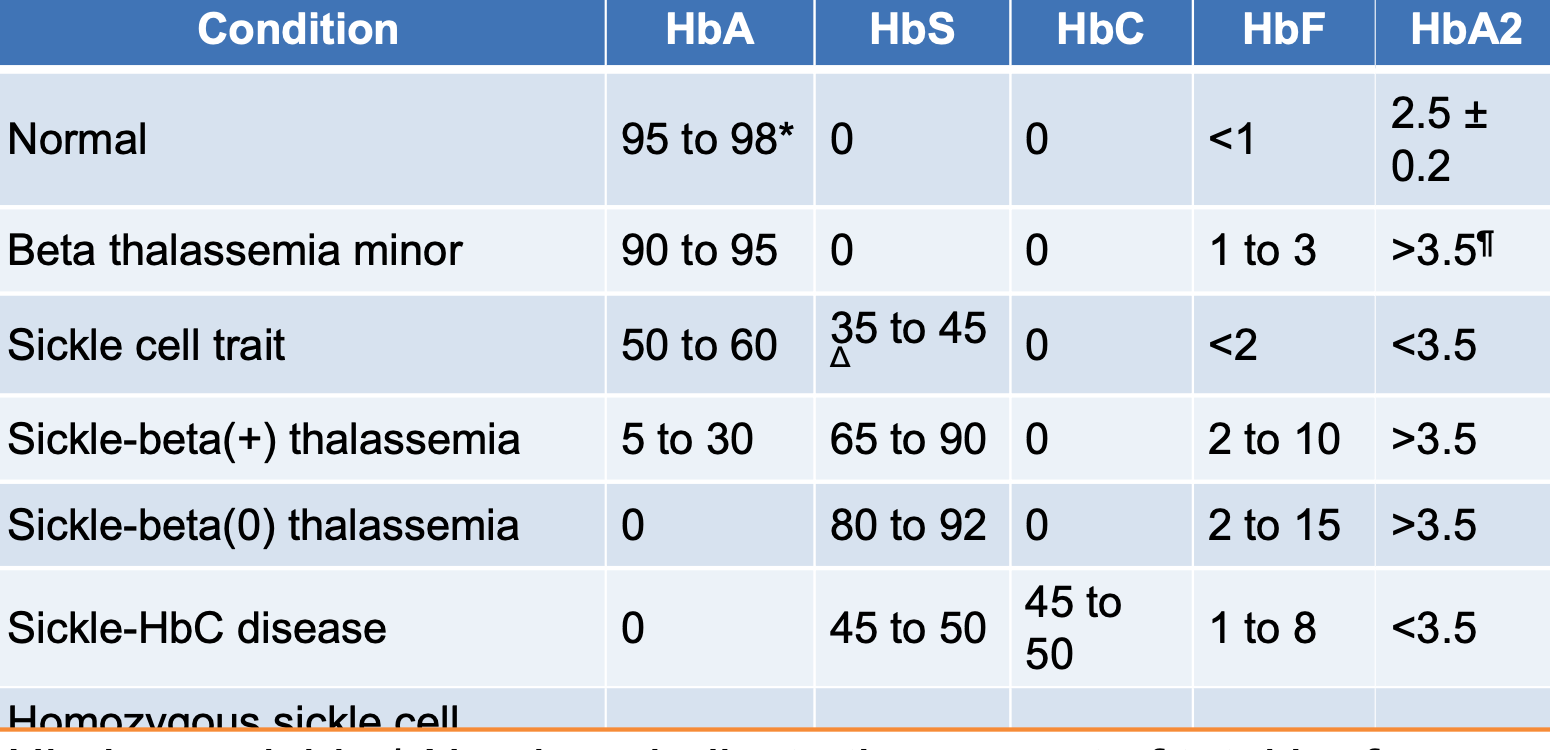
- Hemoglobin electrophoresis: Usually reveals the diagnosis. Normal HbA2 is between 1.5 and 3.0%, while HbA2 >3.5% is diagnostic of β-thalassemia trait.
Other Tests
- DNA analysis: Offered to confirm couples at risk and for prenatal testing.
- α-thalassemia trait: Requires measuring either the α-β chain synthesis ratio or using polymerase chain reaction (PCR) assay tests.
Laboratory Diagnosis
Electrophoretic Patterns
Imaging
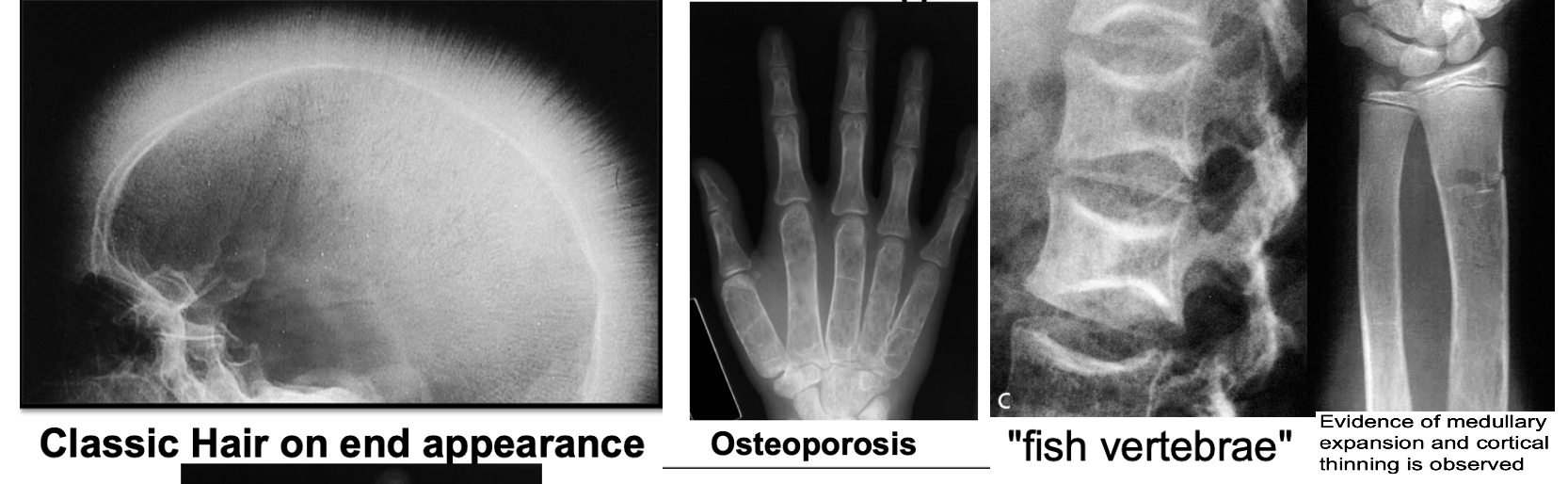
- Skeletal surveys: Show classical changes to the bones.
- Plain skull X-ray: May show the classic “hair-on-end” appearance.
- Generalized bone density loss: Thinned cortex, coarsened trabeculae, and widened medullary cavity.
Management
General Principles
- Asymptomatic carriers: Require no specific treatment but should avoid iron supplementation.
- Occasional blood transfusion: May be necessary.
Thalassemia Intermedia or HbH Disease
- Close monitoring for progression of complications induced by chronic hemolytic anemia.
- Potential complications include:
- Infection-associated aplastic or hyper-hemolytic crises
- Growth impairment
- Skeletal deformities
- Hypersplenism (splenectomy may be considered, but carries risks)
Thalassemia Major
- Regular transfusion: To maintain hemoglobin level above 9.5 g/dL.
- Iron chelation: To prevent overload syndrome.
- Care by multidisciplinary team: Hematologist, specialized nurse, social worker, psychologist, genetic counselor, cardiologist, and liver specialist.
- Splenectomy: May be indicated for hypersplenism.
Non-Drug Management
- Psychological support.
- Genetic counseling.
- Avoid iron-rich foods.
- Extra vitamin E, folic acid, and some vitamin C may be beneficial.
Specific Treatments
- Do not treat anemia with iron unless iron deficiency is confirmed.
- Desferrioxamine: Given parenterally to aid iron excretion.
- Oral chelating agents: Deferasirox and deferiprone.
- Combination therapies: E.g., oral deferiprone with desferrioxamine.
- Hydroxyurea: May increase HbF expression and remove excess α chains, potentially correcting ineffective erythropoiesis.
Thalassemia: Disease Presentation and Complications
Pediatrics
Thalassemia syndromes
Thalassemia syndromes are a heterogenous group of inherited anemias characterized by reduced or absent synthesis of either alpha or Beta globin chains of Hb A.
- Most common single gene disorder
- Autosomal recessive
- Beta thal - point mutations on chromosome 11
BASICS - 3 types of Hb
- Hb A - 2 α and 2 β chains forming a tetramer
- 97% adult Hb
- Postnatal life Hb A replaces Hb F by 6 months
- Fetal Hb – 2α and 2γ (gamma) chains
- 1% of adult Hb
- 70-90% at term. Falls to 25% by 1st month and progressively
- Hb A2 – 2 α and 2 δ chains (delta)
- 1.5 – 3.0% of adult Hb
Classification
-
If synthesis of α chain is suppressed – level of all 3 normal Hb A (2α, 2β), A2 (2α, 2 δ), F (2α, 2γ) reduced – alpha thalassemia
-
Hb H (β4) - Hb-Bart’s (ץ4)
-
If β chain is suppressed - adult Hb is suppressed - beta thalassemia
- β+ thal: reduced synthesis of β globin chain, heterozygous
- β 0 thal: absent synthesis of β globin chain, homozygous ---- Hb A - absent
- Hb F (α2 ץ2)
- Hb A2 (α2 δ2)
Beta Thalassemia
| Syndrome | Clinical Features | Hemoglobin Pattern | β Globin Genes Affected |
|---|---|---|---|
| Silent carrier | Asymptomatic, no anemia, normal | Normal, diagnosed by chain synthesis | Heterozygous state |
| Thalassemia trait | Mild anemia, hypochromic microcytic red cells | Elevated HbA2 > 3.4% | Heterozygous state |
| Thalassemia intermedia | Moderate; not dependent on blood transfusion for their survival. May require some transfusion | HbF elevated | Homozygous state |
| Thalassemia major | Dependent on blood transfusion for their survival. Requires regular transfusion | HbF markedly elevated | Homozygous state |
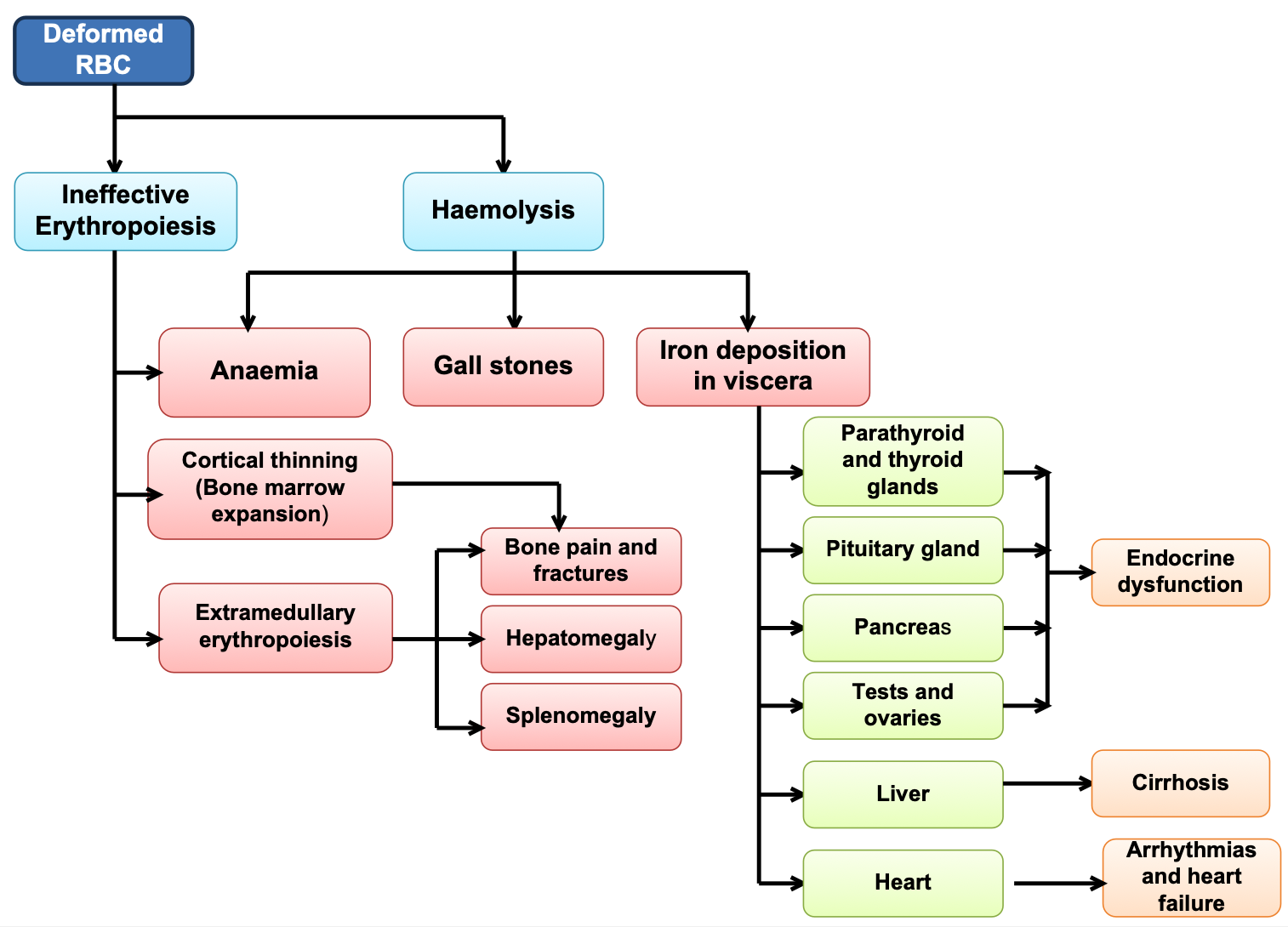
EFFECTS OF MARROW EXPANSION
- Pathological fractures due to cortical thinning
- Deformities of skull and face
- Sinus and middle ear infection due to ineffective drainage
- Folate deficiency
- Hypermetabolic state → fever, wasting
- Increased absorption of iron from intestine
JAUNDICE
- Unconjugated hyperbilirubinemia - hemolysis
- Hepatitis - transfusion, hemochromatosis
- GB stones - obstructive jaundice
- cholangitis
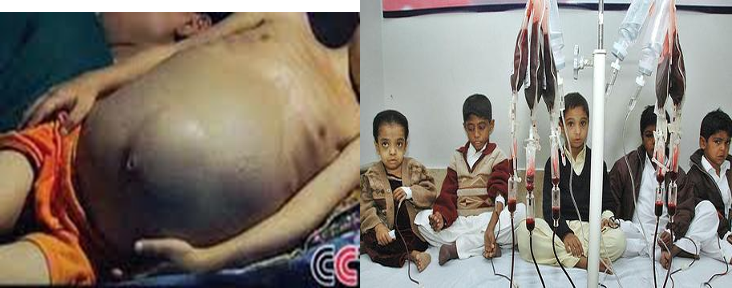
HEPATOMEGALY
- Extra medullary erythropoiesis
- Iron released from breakdown of endogenous or transfused RBCs cannot be utilized for Hb synthesis – hemosiderosis
- Hemochromatosis
- Infections – transfusion related - Hep B, C, HIV
- Chronic active hepatitis
SPLENOMEGALY
- Extra medullary hematopoiesis
- Work hypertrophy due to constant hemolysis
- Hypersplenism (progressive splenomegaly)
INFECTIONS
- Poor nutrition
- Increased iron in body
- Blockage of monocyte-macrophage system
- Hypersplenism - leukopenia
- Infections associated with transfusions
ACCUMULATION OF IRON
- Deposition in pituitary - endocrine disturbance, short stature, delayed puberty, poor secondary sexual characteristics
- Hemochromatosis - cirrhosis of liver
- Cardiomyopathy (cardiac hemosiderosis) - cardiac failure, sterile pericarditis, arrhythmias, heart block
- Deposition in pancreas - diabetes mellitus
- Lungs: restrictive lung defects
- Adrenal insufficiency
- Hypothyroidism, hypoparathyroidism
- Increased susceptibility to infections (iron favors bacterial growth) especially: Yersinia infections
Clinical manifestations of Thalassemia Major
INFANTS:
- Age of presentation: 6-9 mo (Hb F replaced by Hb A)
- Progressive pallor and jaundice
- Cardiac failure
- Failure to thrive, gross motor delay
- Feeding problems
- Bouts of fever and diarrhea
- Hepatosplenomegaly
BY CHILDHOOD:
- Growth retardation
- Severe anemia - cardiac dilatation
- Transfusion dependent
- Icterus
- Changes in skeletal system
SKELETAL CHANGES
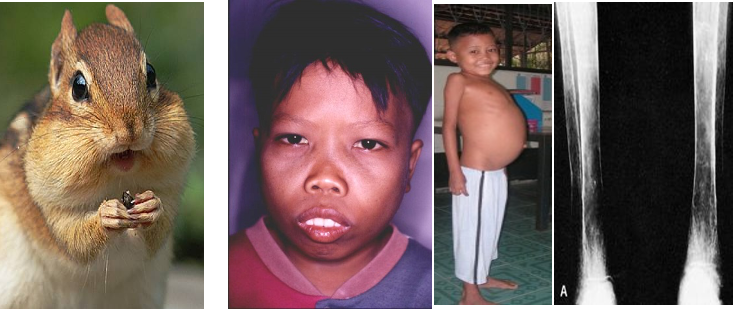
- CHIPMUNK FACIES (HEMOLYTIC FACIES):
- Frontal bossing, maxillary hypertrophy,
- Depression of nasal bridge, Malocclusion of teeth
- PARAVERTEBRAL MASSES:
- Broad expansion of ribs at vertebral attachment
- Paraparesis
- PATHOLOGICAL FRACTURES:
- Cortical thinning (in long bones)
- Increased porosity of long bones
- DELAYED PNEUMATISATION OF SINUSES
- PREMATURE FUSION OF EPIPHYSES - Short stature
Other changes
- Delayed menarche
- Gall-stones, leg ulcers
- Evidence of hypersplenism
Clinical Features of Thalassemia Intermedia
- Moderate pallor, usually maintains Hb >6gm%
- Anemia worsens with pregnancy and infections (erythroid stress)
- Less transfusion dependent
- Skeletal changes present, progressive splenomegaly
- Growth retardation
- Longer survival than Thal major
Clinical Features Of Thalassemia Minor
- Usually ASYMPTOMATIC
- Mild pallor, no jaundice
- No growth retardation, no skeletal abnormalities, no splenomegaly
- MAY PRESENT AS IRON DEFICIENCY ANEMIA (Hypochromic microcytic anemia)
- Unresponsive/refractory to Fe therapy
- Normal life expectancy
Hb
Lab Findings and investigations
- Hb Electrophoresis
| Beta Thalassemia Major | Beta Thalassemia Intermedia | Beta Thalassemia Minor | |
|---|---|---|---|
| HbF | 90-96% | 20-100% | 1-5% |
| HbA2 | 3.5%-5.5% | 3.5%-5.5% | 3.5%-7.5% |
| HbA | 0% | 0-30% | 80-95% |
Hb A- 97% adult Hb (2α and 2β chains)
- Postnatal life Hb A replaces Hb F by 6 months
Hb F -1% of adult Hb (2α and 2γ chains)
- 70-90% at term. Falls to 25% by 1st month and progressively
Hb A2- 1.5 – 3.0% of adult Hb (2 α and 2 δ chains)
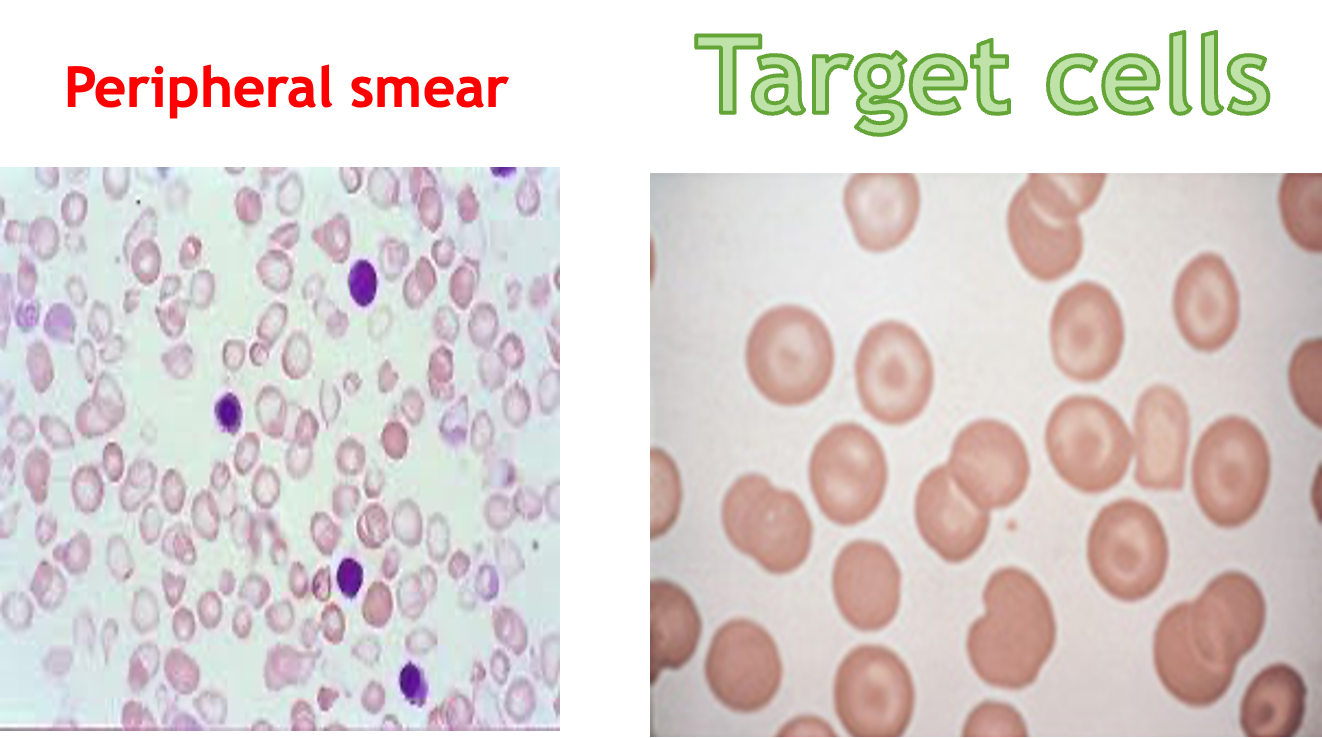
Peripheral smear
- Peripheral blood film shows – anemia, high reticulocytes, numerous nucleated cell, microcytosis
- Target cells
- Unconjugated bilirubin - increased
- Serum ferritin and transferrin – elevated
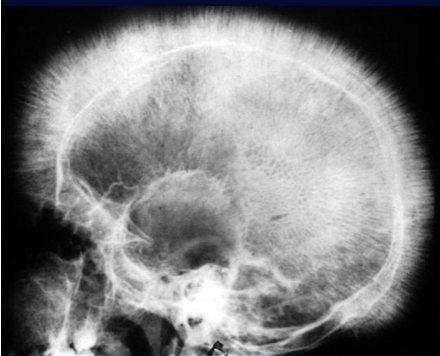
- X-ray - bone marrow hyperplasia, Skull – interrupted porosity gives hair on end appearance
- DNA analysis - confirm, prenatal diagnosis, detection of carrier
- Liver function test
- Infection screen - HIV, Hepatitis B and C, VDRL (before first transfusion)
- HLA typing (for all patients with unaffected siblings)
Treatment
-
Bone marrow transplant (cure!)
- HLA-matched siblings
-
Blood transfusion……………… life-long - Goal: Hb – 9.5 g/dl (Post) - Maintain: Hb 12-12.5g/dl and <15.5g/dl (mean post) - Interval: 2-6 weekly interval (mean 4) - Volume: 15-20ml/kg (max) over 4 hours.
-
Hypertransfusion: Maintaining the Hb above a minimum of 10 g/dL.
-
Normotransfusion: Promotes normal growth and development, prevents the onset of severe hepatosplenomegaly and hemolytic facies, lowers the absorption of gastrointestinal iron and reduces the anemia/cardiomyopathy changes
-
-
Supertransfusion:
- Maintain pre-transfusion Hb of above 12 g/dL.
- Moderate transfusion: In Europe, a newer regimen has been adopted and recommended by the Thalassemia International Federation. In this regimen, pretransfusion Hb is maintained between 9 g/dL and 10.5 g/dL.
-
Assess Iron Overload via: - S. Ferritin - Urinary Fe excretion - Liver biopsy - Chemical analysis of tissue Fe - Endomyocardial biopsies - Myocardial MRI indexes - Ventricular function – ECHO, ECG
Iron Chelation Therapy (Prevent hemosiderosis, haemochromatosis)
-
When - > 2 years old and serum ferritin reaches 1000ng/ml (10-20 transfusions)
-
Deferoxamine (Desferal): Aim: Serum ferritin <1000ng/ml. - SC over 10-12 hours via continuous infusion pump - 5-7 days per week (severity dependent)
-
Deferiprone/L1 (Kelfer/Ferriprox) Given: oral
- Dosage: 75-100mg/kg/day in 3 doses/daily
-
Deferasirox (Exjade) - new - Targets: >2 years - Dosage: 20-30mg/kg/day in liquid dispersible tablet (once!)
Surgical Splenectomy
- Indications: a) T. Intermediate - falling steady state Hb
- b) Transfused - Rising transfusion needs
Newer therapies:
-
GENE MANIPULATION AND REPLACEMENT - Remove defective β gene and stimulate γ gene - 5-azacytidine increases γ gene synthesis
-
Hb F AUGMENTATION - Hydroxyurea - Myelaran - Butyrate derivatives - Erythropoietin in Thal intermedia
Monitoring
Each time
- Clinical assessment: Height, Weight, Liver and Spleen size
- Pre transfusion Hb, platelet, Post transfusion (half hour)
- Volume transfused
Every 3-6 months
- Growth & Development
- Serum Ferritin
- LFT
Every year (/more)
- Growth & Development
- Endocrine assessment (RBS, T4/TSH, Ca(PTH and vit D, Po4)
- Pubertal and Sexual (>10 years)
- FSH, LH, Estrogen and testosterone
- Infection screen (Hep B and C, HIV, VDRL) (6 monthly)
- Cardiac assessment - ECG, Echo
- Liver Iron store
IDA vs THALASSEMIA
-
Iron deficiency anaemia
- Low ferritin.
- High TIBC.
- Normal or low retics.
- High RDW
-
Thalassemia
- Normal or high ferritin.
- Normal or low TIBC.
- High retics.
- Normal RDW
The following findings are noted in individuals with alpha thalassemia trait:
- Asymptomatic/mild anemia
- Hemoglobin level - Within the reference range
- Reticulocyte count - Normal
- MCV - 65-75 fL (80–100 fl)
- MCH - Around 22 pg (27 to 31)
FM
- Inherited autosomal recessive trait.
- Of two types:
- Minor &
- Major (Cooley’s anemia). Thalassemia major has been called Mediterranean anemia.
Clinical Presentation
Hb Electrophoresis
Alpha thalassemia
- 1 gene deletion: normal patient.
- 2 gene deletion: mild anemia with hematocrit 30–40% and low MCV.
- 3 gene deletion: profound anemia with hematocrit 22–32% and very low MCV.
- 4 gene deletion: die in utero.
Beta thalassemia
- Trait: mild anemia with marked microcytosis (low MCV).
- Cooley’s anemia: severely symptomatic age 6 months.
- growth failure, hepatosplenomegaly, jaundice, and bony deformities secondary to extramedullary hematopoiesis.
- They are later symptomatic from hemochromatosis, cirrhosis, and CHF from chronic anemia and transfusion dependence.
Thalassemia Signs and Symptoms
- Weakness, fatigue, and malaise
- Pallor
- Deformities of facial bones
- Slow Growth
- Urine is dark and concentrated
- Abdominal Swelling
- Massive bone marrow expansion occurs

Diagnosis of Thalassemia
- Family history
- CBC: Smaller sized red blood cells (MCV) as well as low RBC counts.
- Iron studies
- Ferritin level
- Liver biopsy
Electrophoresis Result in Thalassemia
- If thalassemia is still suspected, a haemoglobin electrophoresis may help diagnose the condition. The haemoglobin electrophoresis with beta thalassemia trait usually has:
- Reduced or absent HbA,
- Elevated levels of HbA2, and
- Increased HbF.
Treatment
- Blood transfusion
- Prevent iron overload
- Normal diet
- Splenectomy
Thalassemia Minor (Thalassemia Trait)
-
The individual has only one copy of the beta thalassemia gene (together with one perfectly normal beta-chain gene). Known as carriers.
-
Persons have (at most) mild anemia.
-
This situation can very closely resemble that with mild iron-deficiency anemia. However, persons with thalassemia minor have a normal blood iron level.
-
No treatment is necessary for thalassemia minor.
-
The only way to identify the gene is hemoglobin electrophoresis.
Radiology
z In the skull, there is widening of the diploë and there may be perpendicular striations giving an appearance known as ‘hair-on-end’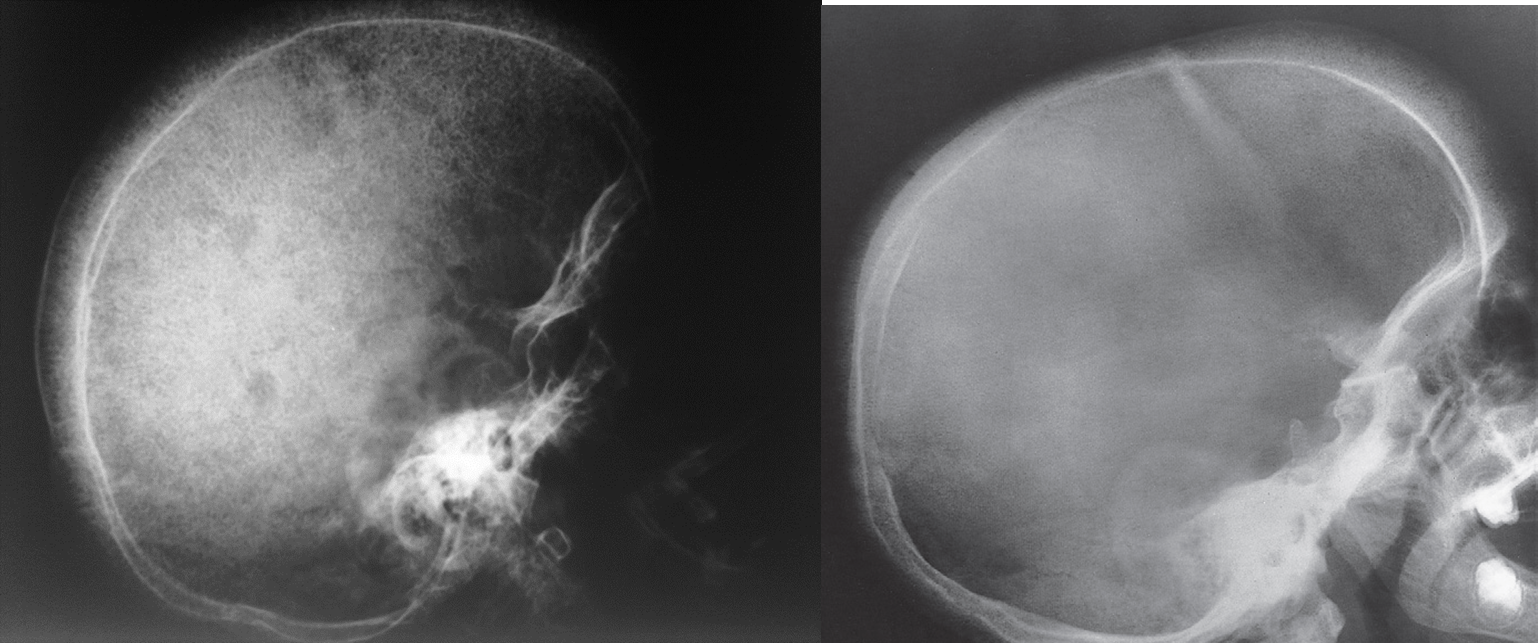
Hair-on-end appearance of the skull
X-ray skull (lateral view) of a child with beta thalassemia Expansion of the diploic space with thickening of the trabeculae is the result of extramedullary hematopoiesis. The hair-on-end appearance produced can also be seen in other chronic hemolytic anemias. e.g. Sickle cell Disease
Thalassaemia haemolytic anaemia. Skull showing thickened diploë