Sarcoidosis Overview
Sarcoidosis is a systemic inflammatory disease characterized by the formation of non-caseating granulomas in various organs, most commonly the lungs and lymphatic system. The exact cause of sarcoidosis is unknown, but it is believed to result from an exaggerated immune response to an unknown antigen in genetically predisposed individuals.
Epidemiology
- Prevalence: Sarcoidosis is more common in certain populations, particularly African Americans and Northern Europeans.
- Age: It typically affects adults between the ages of 20 and 40.
- Gender: Slightly more common in women than in men.
Etiology
- Genetic Factors: Certain genetic markers, such as HLA-DRB1 and BTNL2, have been associated with an increased risk of sarcoidosis.
- Environmental Triggers: Potential triggers include infectious agents (e.g., mycobacteria, Propionibacterium acnes), occupational exposures (e.g., beryllium, silica), and other environmental factors.
- Immune Response: The disease is thought to involve an exaggerated Th1 immune response, leading to granuloma formation.
Pathophysiology
- Granuloma Formation: The hallmark of sarcoidosis is the formation of non-caseating granulomas, which are organized collections of macrophages, epithelioid cells, and multinucleated giant cells surrounded by lymphocytes.
- Immune Dysregulation: The granulomas form as a result of an exaggerated immune response, particularly involving CD4+ T-helper cells, which release cytokines like IFN-γ and TNF-α that promote macrophage activation and granuloma formation.
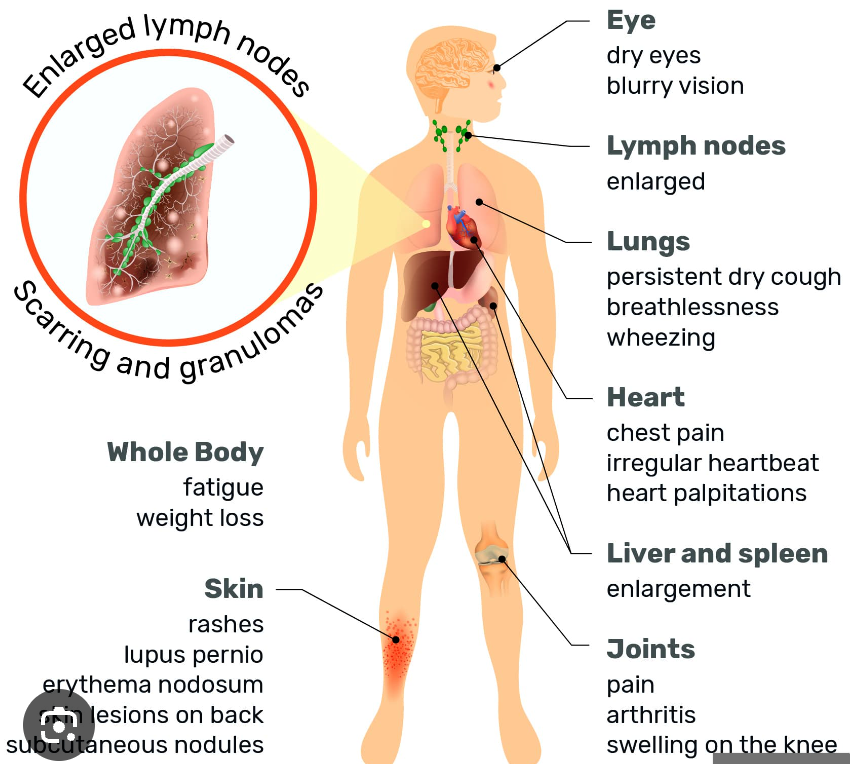
- Organ Involvement: Granulomas can form in virtually any organ, but the lungs and lymph nodes are most commonly affected. Other organs that may be involved include the skin, eyes, liver, spleen, heart, and nervous system.
Clinical Manifestations
-
Pulmonary Sarcoidosis:
- Symptoms: Cough, dyspnea, chest pain, and wheezing.
- Radiographic Findings: Bilateral hilar lymphadenopathy, interstitial lung disease, and pulmonary nodules.
- Stages: Classified into four stages based on chest X-ray findings:
- Stage I: Bilateral hilar lymphadenopathy without lung involvement.
- Stage II: Bilateral hilar lymphadenopathy with pulmonary infiltrates.
- Stage III: Pulmonary infiltrates without hilar lymphadenopathy.
- Stage IV: Pulmonary fibrosis.
-
Extrapulmonary Sarcoidosis:
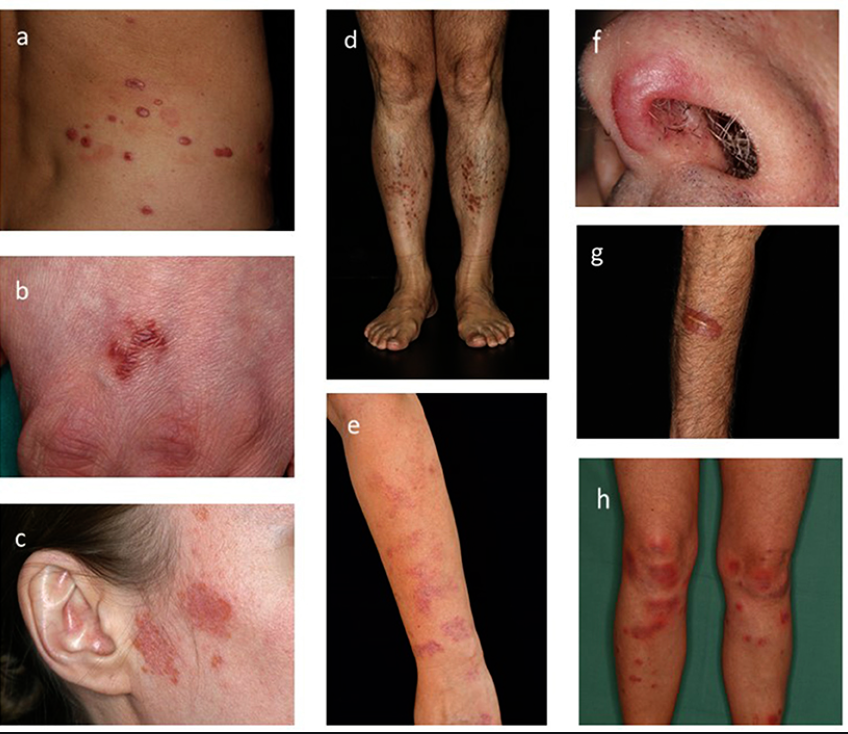
- Skin: Erythema nodosum, lupus pernio, maculopapular rash.
- Eyes: Uveitis, conjunctivitis, keratoconjunctivitis sicca.
- Cardiac: Arrhythmias, heart block, cardiomyopathy.
- Neurological: Cranial nerve palsies (especially facial nerve), meningitis, peripheral neuropathy.
- Liver and Spleen: Hepatomegaly, splenomegaly, elevated liver enzymes.
- Musculoskeletal: Arthralgia, arthritis, myopathy.
-
Systemic Symptoms: Fatigue, fever, weight loss, night sweats.
Diagnosis
- Clinical Evaluation: Diagnosis is based on clinical presentation, radiographic findings, and exclusion of other granulomatous diseases.
- Imaging:
- Chest X-ray: Often the first imaging study performed; shows bilateral hilar lymphadenopathy.
- High-Resolution CT (HRCT): Provides more detailed images of lung parenchyma and is useful in assessing the extent of lung involvement.
- PET Scan: May be used to assess active inflammation and guide biopsy.
- Laboratory Tests:
- Serum Angiotensin-Converting Enzyme (ACE): Elevated in about 60% of patients but not specific.
- Hypercalcemia/Hypercalciuria: Due to increased production of 1,25-dihydroxyvitamin D by activated macrophages in granulomas.
- Elevated Inflammatory Markers: ESR, CRP.
- Biopsy: The definitive diagnosis is made by histological examination of tissue showing non-caseating granulomas. Common biopsy sites include the lung, lymph nodes, skin, and transbronchial biopsy.
- Pulmonary Function Tests (PFTs): May show restrictive lung disease with reduced lung volumes and diffusing capacity (DLCO).
Differential Diagnosis
- Infectious Granulomatous Diseases: Tuberculosis, fungal infections (e.g., histoplasmosis, coccidioidomycosis).
- Other Granulomatous Diseases: Granulomatosis with polyangiitis, berylliosis, hypersensitivity pneumonitis.
- Malignancy: Lymphoma, metastatic cancer.
Treatment
- Observation: Many cases of sarcoidosis are self-limiting and do not require treatment.
- Corticosteroids: The mainstay of treatment for symptomatic sarcoidosis. Prednisone is commonly used.
- Steroid-Sparing Agents: For patients who require long-term treatment or cannot tolerate corticosteroids, alternatives include:
- Methotrexate
- Azathioprine
- Mycophenolate mofetil
- Hydroxychloroquine: Particularly useful for skin and joint involvement.
- Biologic Agents: Anti-TNF agents (e.g., infliximab) may be used in refractory cases.
- Organ-Specific Management:
- Pulmonary: Inhaled corticosteroids may be used for mild disease; oxygen therapy for advanced disease.
- Cardiac: Pacemaker or defibrillator for arrhythmias; heart transplant in severe cases.
- Ocular: Topical corticosteroids for anterior uveitis; systemic therapy for posterior uveitis or optic neuritis.
Prognosis
- Variable Course: Sarcoidosis can be acute and self-limiting or chronic and progressive.
- Spontaneous Remission: Occurs in about 60-70% of cases, particularly in those with Stage I disease.
- Chronic Disease: About 10-30% of patients develop chronic sarcoidosis, which may lead to organ damage and complications.
- Mortality: Rare, but can occur due to complications such as pulmonary fibrosis, cardiac involvement, or neurosarcoidosis.
Complications
- Pulmonary Fibrosis: Can lead to respiratory failure.
- Pulmonary Hypertension: Secondary to chronic lung disease.
- Cardiac Sarcoidosis: Can cause arrhythmias, heart block, or heart failure.
- Neurosarcoidosis: Can lead to cranial nerve palsies, meningitis, or seizures.
- Hypercalcemia: Can lead to nephrolithiasis and renal failure.
Monitoring and Follow-Up
- Regular Monitoring: Patients with sarcoidosis should be regularly monitored for disease progression and treatment side effects.
- Pulmonary Function Tests: To assess lung function over time.
- Imaging: Periodic chest X-rays or HRCT to monitor lung involvement.
- Laboratory Tests: Monitoring of serum calcium, liver function tests, and ACE levels.
Summary
Sarcoidosis is a complex, multisystem disease with a variable clinical course. Diagnosis is based on clinical presentation, imaging, and biopsy showing non-caseating granulomas. Treatment is tailored to the severity of the disease and the organs involved, with corticosteroids being the mainstay of therapy. Regular monitoring is essential to manage the disease and prevent complications.