Cystic Fibrosis
Background
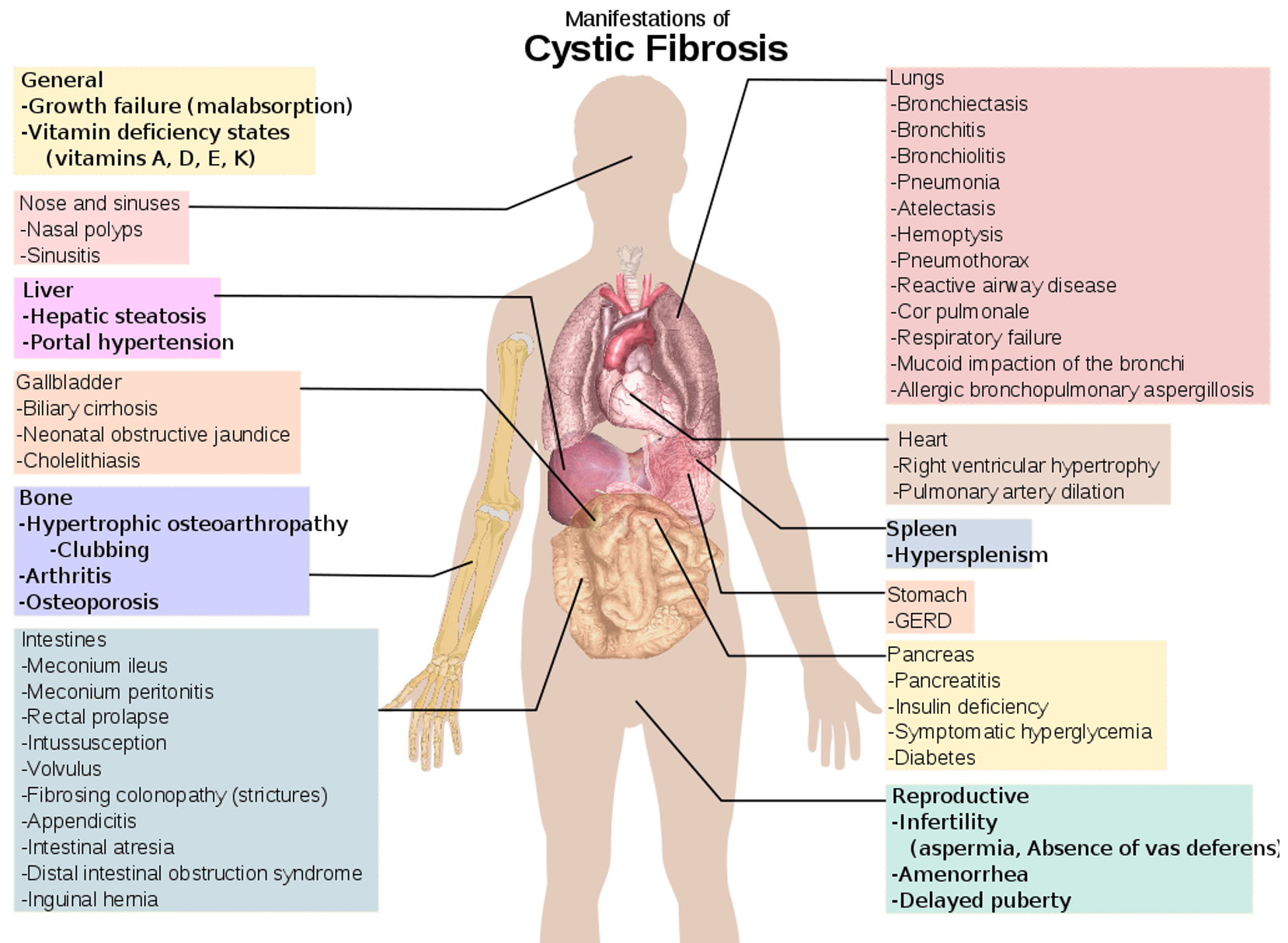
Cystic fibrosis (CF) is a disease of exocrine gland function that involves multiple organ systems but chiefly results in chronic respiratory infections, pancreatic enzyme insufficiency, and associated complications.
Pathophysiology
-
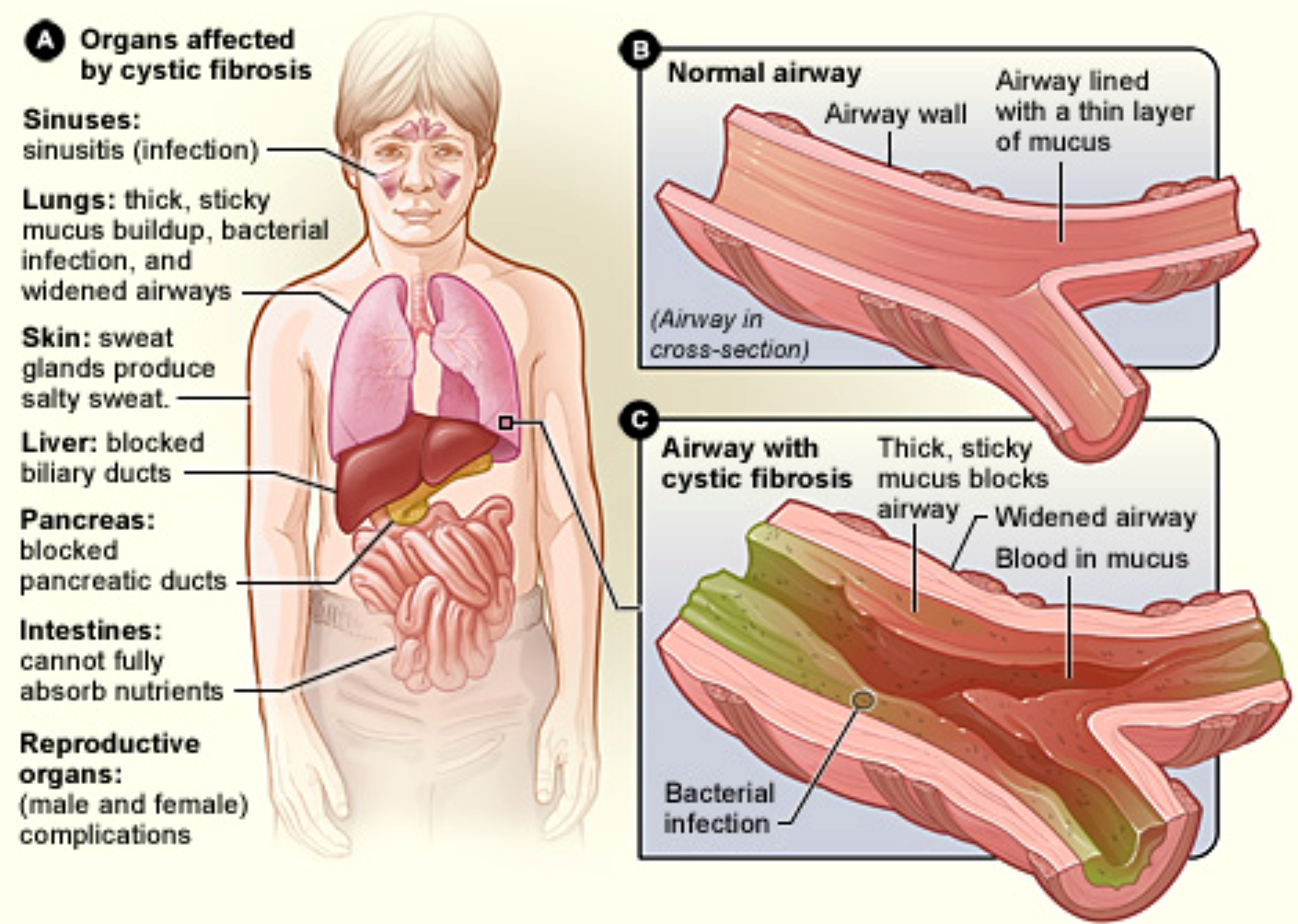
Cystic fibrosis is caused by defects in the CF gene, which codes for a protein transmembrane conductance regulator (CFTR).
-
CFTR functions as a chloride channel and is regulated by cAMP.
-
Mutations in the CFTR gene result in abnormalities of cAMP-regulated chloride transport across epithelial cells on mucosal surfaces.
-
Cystic fibrosis is an autosomal recessive disease.
-
Its estimated heterozygote frequency in white people is up to 1 in 20. Median survival age is 36.9 years.
History
- Median age at diagnosis of cystic fibrosis is 6-8 months.
- Approximately 10% of patients with cystic fibrosis remain pancreatic sufficient; these patients tend to have a milder course.
Neonates: may present with meconium ileus or, rarely, anasarca.
Patients younger than 1 year present with:
- wheezing, coughing, recurring respiratory infections, pneumonia,
- steatorrhea, failure to thrive, or both.
Patients diagnosed later in childhood:
- are more likely to have pancreatic sufficiency and often present with chronic cough and sputum production.
Meconium ileus
occurs in 7-10% of patients with CF.
-
Simple meconium ileus presents with abdominal distension at birth, progressing to failure to pass meconium, bilious vomiting, and progressive abdominal distension.
-
Complicated meconium ileus presents more dramatically at birth with severe abdominal distention, accompanied by abdominal wall erythema and edema.
- Intestinal obstruction at birth (e.g., volvulus, intestinal atresia, perforation, meconium peritonitis).
- Delayed passage of meconium (>24-48 hours after birth).
- Prolonged cholestatic jaundice.
Pancreatic insufficiency:
- Fat-soluble vitamin deficiency and malabsorption of fats, proteins, and carbohydrates.
- Malabsorption results in steatorrhea, characterized by frequent, poorly formed, large, bulky, foul-smelling, greasy stools that float in water. Some patients have anorexia without obvious steatorrhea.
- Failure to thrive, flatulence or foul-smelling flatus, recurrent abdominal pain, and abdominal distention.
- Intussusception (ileocecal).
- Rectal prolapse.
- Jaundice or GIT bleeding as a result of hepatobiliary involvement.
Respiratory Tract Manifestations
- Chronic or recurrent cough: dry and hacking at the beginning and produce mucoid (early) and purulent (later) sputum. - psuodomonas most common
- Prolonged symptoms of bronchiolitis in infants.
- Paroxysmal cough followed by vomiting.
- Recurrent wheezing, recurrent pneumonia, atypical asthma, pneumothorax, hemoptysis, and digital clubbing are all complications.
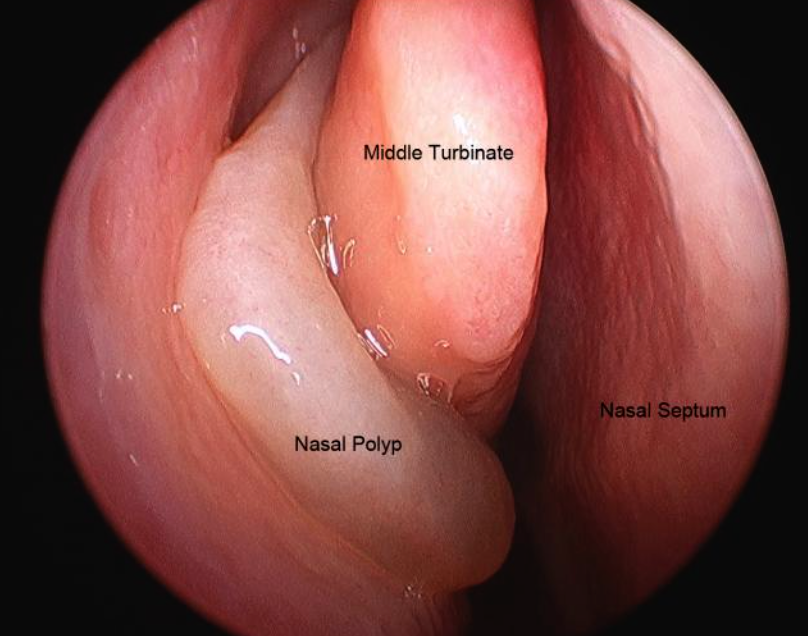
- Dyspnea on exertion, history of chest pain, recurrent sinusitis, nasal polyps; Sinusitis, and hemoptysis.
Urogenital Tract Manifestations
- Undescended testicles or hydrocele.
- Males are frequently sterile because of the absence of the vas deferens.
- In females, decreased fertility, secondary sexual development is often delayed.
- Amenorrhea may occur in females with severe nutritional or pulmonary involvement.
Physical Examination - Respiratory System
-
Rhinitis
-
Nasal polyps
-
Tachypnea
-
Respiratory distress with retractions
-
Wheeze or crackles
-
Cough (dry or productive of mucoid or purulent sputum)
-
Increased anteroposterior diameter of chest
-
Clubbing
-
Cyanosis
-
Hyperresonant chest upon percussion
-
Crackles are heard acutely in associated pneumonitis or bronchitis and chronically with bronchiectasis.
-
Swelling of submandibular gland or parotid gland.
-
Abdominal distention.
-
Hepatosplenomegaly (fatty liver and portal hypertension).
-
Rectal prolapse.
-
Dry skin (vitamin A deficiency).
-
Cheilosis (vitamin B complex deficiency).
-
Scoliosis/Kyphosis.
Differential Diagnoses
- Acute Sinusitis
- Bronchiolitis
- Failure to Thrive
- Asthma
- Bronchiectasis
- Celiac Disease (Sprue)
- Primary Ciliary Dyskinesia
- Short Stature
Diagnostic Confirmation
Requirements for a CF diagnosis include either positive genetic testing or positive sweat chloride test findings (>60 mEq/L) and one of the following:
- Typical chronic obstructive pulmonary disease
- Documented exocrine pancreatic insufficiency
- Positive family history (usually affected sibling).
Prenatal Testing:
- Noninvasive CFTR analysis involves a technique for recovering DNA from cells obtained by buccal brushing.
- Amniocentesis.
Neonatal Testing:
- Immunoreactive trypsinogen (IRT) elevated in infants with cystic fibrosis and genetic testing.
Other Tests for CF
- Comprehensive metabolic panel (CMP): Urea, electrolytes, Ca, LFT.
- Complete blood count (CBC)—another test for general health and to help detect infections.
- Glucose and hemoglobin A1c—to detect, diagnose, and monitor diabetes.
- Amylase and lipase—to detect problems with the pancreas.
- Fecal fat—to identify malabsorption and pancreatic insufficiency.
- Semen analysis—to detect infertility.
- Sputum cultures—to help diagnose lung infections.
- Chest X-rays, upper GI and small bowel series, and lung function tests.
Treatment
The primary goals of CF treatment include the following:
-
Maintaining lung function as near to normal as possible by controlling respiratory infection and clearing airways of mucus.
-
Administering nutritional therapy (i.e., enzyme supplements, multivitamin and mineral supplements) to maintain adequate growth.
-
Managing complications.