Internal Medicine
DR WAQAR
NORMAL
- RBCs are round
- Most of the hemoglobin is Hb.A
WHAT IS SICKLE CELL DISEASE
- It is a type of hemoglobinopathy
- It is a hereditary condition
- An abnormal Hb. called Hb.S, replaces the normal Hb.A
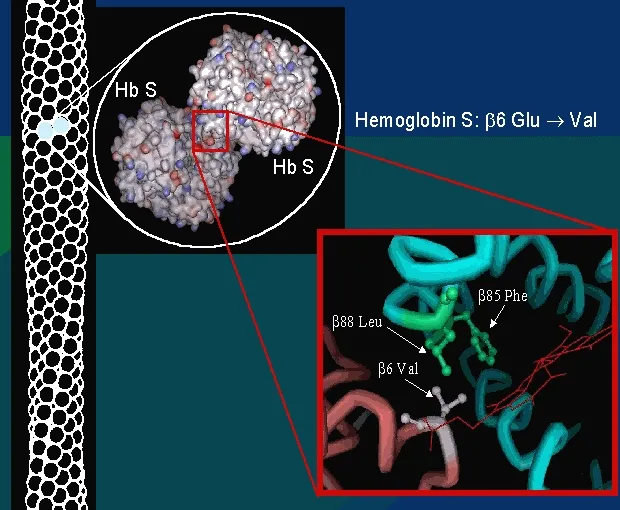
- Hb.S is produced when the amino acid “glutamic acid” (in the normal Hb.A) is replaced by another amino acid “valine”
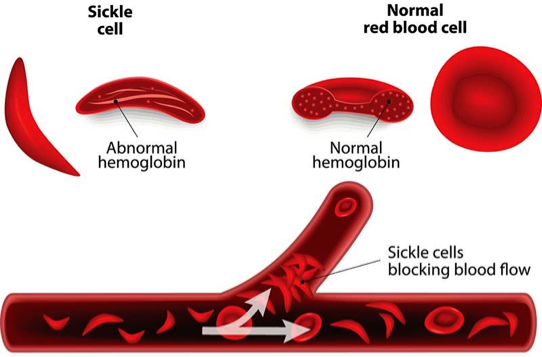
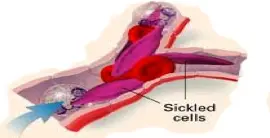
- The Hb.S causes the RBCs to become sickle-shaped (MANJAL) which causes problems (blocking of blood vessels & hemolysis)
VASO-OCCLUSION BY SICKLED CELLS
GENETICS OF SCD
- Pattern of inheritance: Autosomal recessive
- Normally, Hb. is produced by 2 genes
- If both genes are abnormal → sickle cell disease (no Hb. A)
- If only one gene is abnormal, it is called sickle cell trait (carrier state, Hb.AS) → half of body’s Hb is HbS & half is Hb A. Not full-blown disease
- Normal Genetics: AA
- Sickle cell trait: AS
- Sickle cell disease: SS
VARIANTS OF SCD
- Hb.SC disease: Presence of Hb.S plus Hb.C (hemoglobin C is another type of abnormal Hb.)
- Hb.S/β: Sickle cell gene plus beta-thalassemia gene
- Sickle cell disease (Hb.SS) → Symptomatic
- Sickle cell trait (Hb.AS) → usually asymptomatic but can cause sudden death if patients do vigorous exercise
- Hb.S/β disease (sickle + thalassemia) → Symptomatic
- Hb.SC disease: Symptomatic, but milder than SCD
EPIDEMIOLOGY
- Very common worldwide
- In the U.S., 1 in 625 births have SCD
- More in African-Americans
- Common in Mideast, Africa, Mediterranean areas (Greece, Turkey, Italy)
- In KSA, high prevalence in Eastern region & southwestern areas (Jizan)
SIGNS & SYMPTOMS
A stable patient (no acute symptoms) will have:
- Anemia (Hb. around 8)
- May be jaundice (due to?)
- Sometimes leg ulcers
Various Crises in SCD
- Vaso-occlusive pain crisis
- Acute chest syndrome
- Aplastic crisis
- Pain Crisis
- Hemolytic crisis
- Splenic sequestration crisis
OTHER FEATURES OF SCD
- Priapism: It is a painful erection of male genitalia. Repeated attacks may cause impotence. Treatment: analgesia, transfusion
- Stroke/TIA: Due to blockage of cerebral vessels.
- Asplenia (non-functioning spleen)
- Repeated vaso-occlusion causes splenic infarcts → spleen shrinks & becomes non-functional
- Most cases occur by the age of 1 year
- Non-functioning spleen → risk of infections with encapsulated bacteria (Pneumococci, meningococci, H.Flu.)
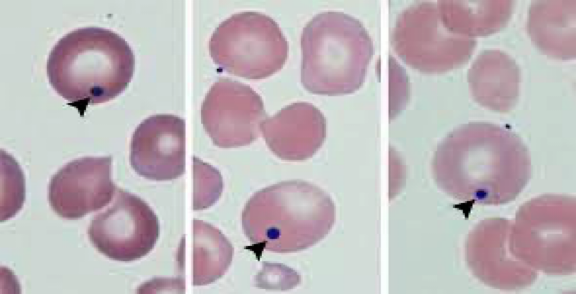
- Howell-Jolly bodies seen on a peripheral smear (remains of DNA in the RBCs) (Howell Jolly bodies indicate asplenia)
(Howell-Jolly bodies seen if there is no spleen)
-
Renal problems:
- Renal medullary infarction → causes tubular damage → impaired water absorption → fluid loss
-
Retinopathy: Due to retinal infarcts (vaso-occlusion)
-
Cholelithiasis: Hemolysis → increased bilirubin → bilirubin stones in the gallbladder (look for a surgical scar)
-
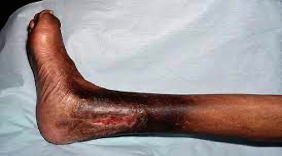
Leg ulcers:
- Due to vaso-occlusion
- Usually on the ankles
-
Pulmonary HTN: Increased risk of this in SCD patients.
-
Bone problems:
- Increased fractures
- Osteomyelitis: Due to Staph Aureus (most common) & Salmonella (most severe)
- Avascular necrosis of femoral & humeral head (due to vaso-occlusion)
-
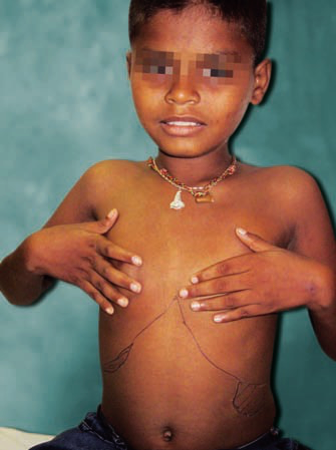
Dactylitis:
- Pain & swelling in the fingers & toes
- Often the presenting feature in children
- Due to ischemic damage to the bones
- Also called HAND-FOOT disease

SCD Investigations
CBC, LFT, Peripheral smear, Electrophoresis, Solubility test, sickling test, genetic testing
Treatments for SCD
Longterm
- Hydroxyurea,
- Folic acid tablets,
- L-Glutamine (oral)
Other Treatments
- Stem cell transplant
- Gene therapy
Rapid Fire Questions
- What is the inheritance pattern of SCD? autosomal recessive
- What is the genotype of a normal person, SCD, and trait? SS fullblown, S trait
- Which amino acid is present in SCD? valine
- Is sickle cell trait symptomatic? usually asymptomatic
- CBC findings in SCD? high plates
- Why is retic count high? hyperactive marrow
- Name some specific tests to diagnose SCD? electrophoresis
- What is the cause of jaundice in SCD? hemolysis
- Name 5 acute crises in SCD? aplastic, splenic, …
- Transfusion is the main treatment of pain crisis, right or wrong? wrong
- What is the cause of pain in acute pain crisis? vasoblockages; ischemic
- 5 indications of transfusion in SCD? HASCS
- What can precipitate a pain crisis? infection, stress
- Treatment of pain crisis? transfusion analgesics
- What is the cause of aplastic crisis? bone marrow suppression due parrovirus b
- What will CBC show in aplastic crisis? everything is low
- Treatment of acute chest syndrome? transfusion, antibiotics, analgesics
- Name 3 crises which can cause a sudden Hb drop? aplastic, hemolytic, splenic…
- What happens to the spleen in SCD patients? fibrosis; aspleenia
- What happens to the eyes, male genitalia, brain, kidneys, bones? retinopathy//occlusions .. .. >
- Patient with SCD has a surgical scar in the right upper abdomen. Why? cholethiasis
- What vaccines should be given to SCD patients? infections that perticipates due asplenia
- In children with SCD, what can happen in the hands & the feet? dactytilitis
- Name 3 medicines which are given long term? folic acid, L-glutamine, Hydroxyurea
- What preventive measures should be taken in SCD patients? prophylaxis, dont go abha, take antibiotics when fever + respiratory distress
- In a patient with SCD, what will you see in a peripheral smear? sickle
- High chances of infection with which bacteria? encapsulated; asplenia
- Name 2 advanced treatments for SCD? stemcells & Gene Therapy
Pediatrics
“Sickle cell disease (SCD) is a term used for a group of genetic disorders characterized by production of Hb “S”. Sickle cell hemoglobinopathy occurs due to mutation of beta-globin gene situated on short arm of chromosome 11, where adenine is replaced by thymine in base of DNA coding for the amino acid in the sixth position in beta-globin chain.
Pathophysiology
-
The “sol” (soluble) form of Hb changes to “gel” form, when Hb “S” is deoxygenated. ⇒
-
In gel form the Hb crystallizes to small, rigid, boat-shaped objects known as ‘’tactoids’’. These tactoids polymerize forming insoluble structure, deforming the RBC to sickle shape. ⇒
-
The RBC membrane becomes more fragile. ⇒
-
The polymerization is facilitated by increased concentration of Hb “S”, acidic pH, low oxygen saturation, stasis of blood flow, decreased levels of Hb”F”.
-
Upon reoxygenation - The sickle cell initially resumes normal configuration, but with repeated cycles of sickling and unsickling, fixation of membrane occurs in sickled configuration, leading to irreversible sickle cell formation, and hemolysis. ⇒
-
Platelets aggregate over the adherent red cells and damaged endothelium, causing blockage of microvasculature and ischemia of the tissue.
-
Infarction of liver, spleen, muscles & bones. (Hepatomegaly & Jaundice, abd. Pain & fibrosis & ↓ spleen size, bone & joint pain).
- Sickle cells sequestered in RE cells and are hemolysed → Anemia
- Chronic Hypoxia → - Clubbing of fingers & toes - Chronic Leg ulcer - Growth retardation
- ↑Bilirubin turnover → Pigment stones
Various Crises in SCD
- Vaso-occlusive pain crisis
- Acute chest syndrome
- Aplastic crisis
- Megaloblastic Crisis
- Pain Crisis
- Hemolytic crisis
- Splenic sequestration crisis
Stroke
- Common age group is 3–10 years. Around 70–90% patients experience repeated episodes of stroke within 36 months unless they are on transfusion regimen maintaining Hb”S” level below 30%.
Priapism
- Priapism is persistent painful penile erection. Recurrent acute episodic attacks last from few minutes to several hours. It usually subsides spontaneously. Impotency may be a sequel. Blood transfusion is indicated if pain, engorgement persists for 24–48 hours.
Clinical Features
- Moderate to severe anemia.
- Mild jaundice.
- Non-healing leg ulcers.
- Decreased physical growth & delayed puberty.
- Clubbing of fingers & toes.
- Enlarged heart and hemic murmur.
- Hepatomegaly.
- Splenomegaly in the beginning.
- Sickle cell crisis –
- Pains & aches.
- ↑ jaundice.
- ↑ anemia.
- Fever.
Complications: Z
- Bones → Dactylytis and chronic osteomyelitis, ⇒ Aseptic necrosis of head of femur.
- Eyes → Sudden blindness.
- Brain → Neurological deficits & emotional disturbances.
Diagnosis Z
- Screening test of older children can be done sickling test.
- Diagnosis can be confirmed by Hb electrophoresis, globin DNA analysis.
- Antenatal diagnosis can be done at 8–10 weeks of gestation, with chorionic villus biopsy, or amniocentesis. DNA analysis using PCR.
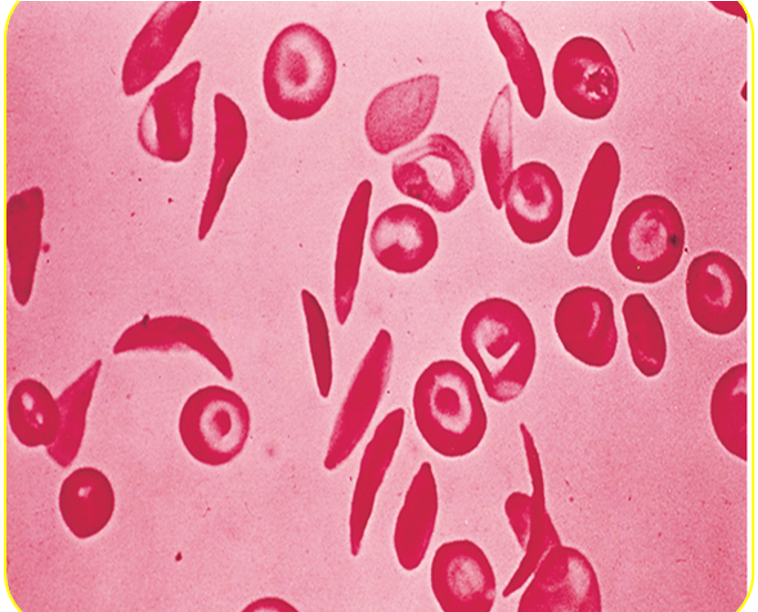
Peripheral smear:
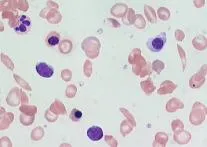
- Deformed sickle-shaped red cells.
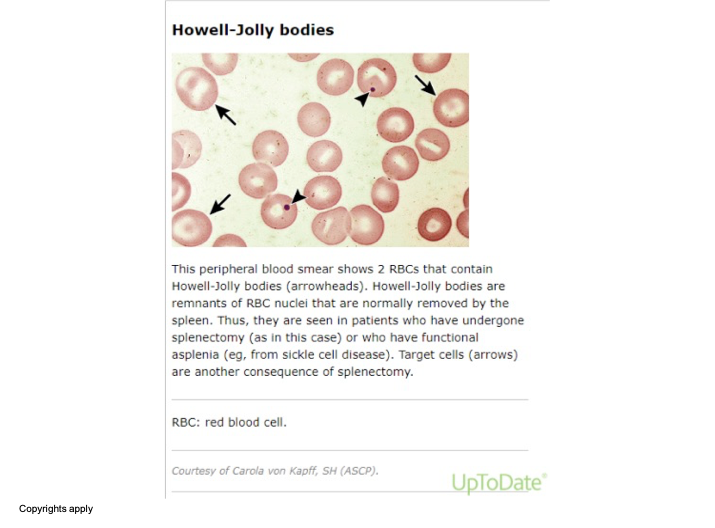 The image shows a microscopic view of a peripheral blood smear. In this image, there are red blood cells (RBCs) with specific annotations pointing out Howell-Jolly bodies (indicated by arrowheads) and target cells (indicated by arrows). Howell-Jolly bodies are remnants of RBC nuclei usually removed by the spleen. They are observed in patients who have undergone a splenectomy or have functional asplenia, such as from sickle cell disease. Target cells can also occur following a splenectomy.
The image shows a microscopic view of a peripheral blood smear. In this image, there are red blood cells (RBCs) with specific annotations pointing out Howell-Jolly bodies (indicated by arrowheads) and target cells (indicated by arrows). Howell-Jolly bodies are remnants of RBC nuclei usually removed by the spleen. They are observed in patients who have undergone a splenectomy or have functional asplenia, such as from sickle cell disease. Target cells can also occur following a splenectomy.
Treatment
Painful Crisis
- Mild pain: codeine, aspirin, ibuprofen, acetaminophen, naproxen.
- Severe pain: meperidine, tolmetin, oxycodone, Ketamine.
- Vasodilators: Pentoxifylline, nifedipine, and buflomedil increase microvascular circulation.
- Hydration.
- Blood transfusion: may be appropriate if pain is accompanied by another complication such as stroke or ACS.
- Hydroxyurea: leads to increase in Hb “F”, which ameliorates the severity of SCD.
Newer Treatments
-
Agents stimulating Hb F - Hydroxy urea - 5 – aza cytidene - Butyrates
-
Agents ↓ Hbs concentration - DDAVP: a synthetic version of antidiuretic hormone (used in von willebrand’s disease to stimulate vWF, may also be used in mild hemophilia A) - Nifedepine, verapamil
-
Membrane active agents – Zinc
-
Hb solubility increasing agents - Cyanate - Urea - Phenyl alanine
A life-long cure for SCD is available only through hematopoietic stem cell transplantation.
Prevention Z
- Avoid precipitating factors for crisis - Exposure to hypoxia, dehydration, extreme cold, extreme heat, change in altitude, infections.
- Avoid Dehydration and loss of electrolytes may lead to red cell dehydration precipitating sickling process.
- Parent’s education.
- Immunization: pneumococcal and Influenzae (Hib) vaccine. Hepatitis B vaccine.
- Hemoglobin levels should be maintained at 10–12 g/dl.
- Folate supplementation.
- Genetic counseling and mass education of high-risk.
- Oral penicillin prophylaxis. Oral penicillin should be given to children up to the age of 5 years.
FM
Jazan Najran Z
Sickle Cell Anemia
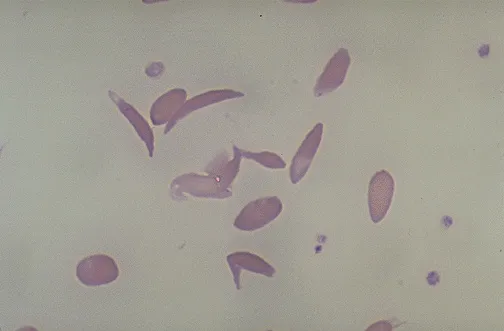
Sickle Cell Anemia
- An inherited disease of red blood cells
- Affects hemoglobin
- Polymerization of hemoglobin leads to a cascade of effects decreasing blood flow
- Tissue hypoxia causes acute and chronic damage
Why Do Cells Sickle?
- Glutamic acid is substituted for valine
- Allowing the polymerization of sickle hemoglobin when deoxygenated
Normal Vs. Sickle Red Cells
Normal
- Disc-Shaped
- Deformable
- Life span of 120 days
Sickle
- Sickle-Shaped
- Rigid
- Lives for 20 days or less
Hematology Overview
Pathology
Hemolysis
-
The anemia in SCD is caused by red cell destruction, or hemolysis, and the degree of anemia varies widely between patients.
-
The production of red cells by the bone marrow increases dramatically, but is unable to keep pace with the destruction.
Vaso-occlusion
-
Occurs when the rigid sickle-shaped cells fail to move through the small blood vessels, blocking local blood flow to a microscopic region of tissue.
-
Amplified many times, these episodes produce tissue hypoxia.
-
The result is pain, and often damage to organs.
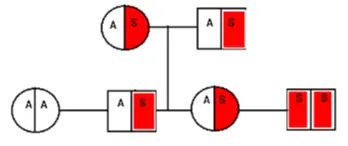
Sickle Cell Pedigree
- Parents with sickle cell trait: hemoglobin AS
- Probability of child with hemoglobin AA: 25%
- Probability of child with sickle cell trait AS: 50%
- Probability of child with sickle cell disease SS: 25%
Clinical Manifestations
- Thrombotic crises
- Aplastic crises
- Hemolytic crises
- Sequestration crises
Laboratory Findings
- Anaemia and unconjugated hyperbilirubinemia
- Sickling test: positive
- Hb electrophoresis: Hbs and HbA2 and HbF but not HbA
Treatment
- Blood transfusion in severe cases and aplastic crises
- Infection prevention
- Pain management
- Maintenance of hydration
- Chronic disease management
- Splenectomy if the spleen is huge

Radiology
Acute clinical manifestations
- Vaso-occlusive events
- Dactylitis in children < 5 years of age
- Vasoocclusive crises (sickle cell pain crisis) Acute chest syndrome
- Organ infarctions (any organ; particularly the spleen) It is well demonstrated on CT
- Avascular necrosis
Infection
- Pneumonia
- Osteomyelitis (most common cause: Salmonella;Staph Aureus)
- Acute hemolytic crisis
MSK imaging features of SCD:
-
Bone infarction: Occur in the intra-articular portions of the bones (avascular necrosis ) or in the medulla of a bone
-
Hair-on-end sign
-
Evidence of bone infection.
-
H-shaped vertebrae
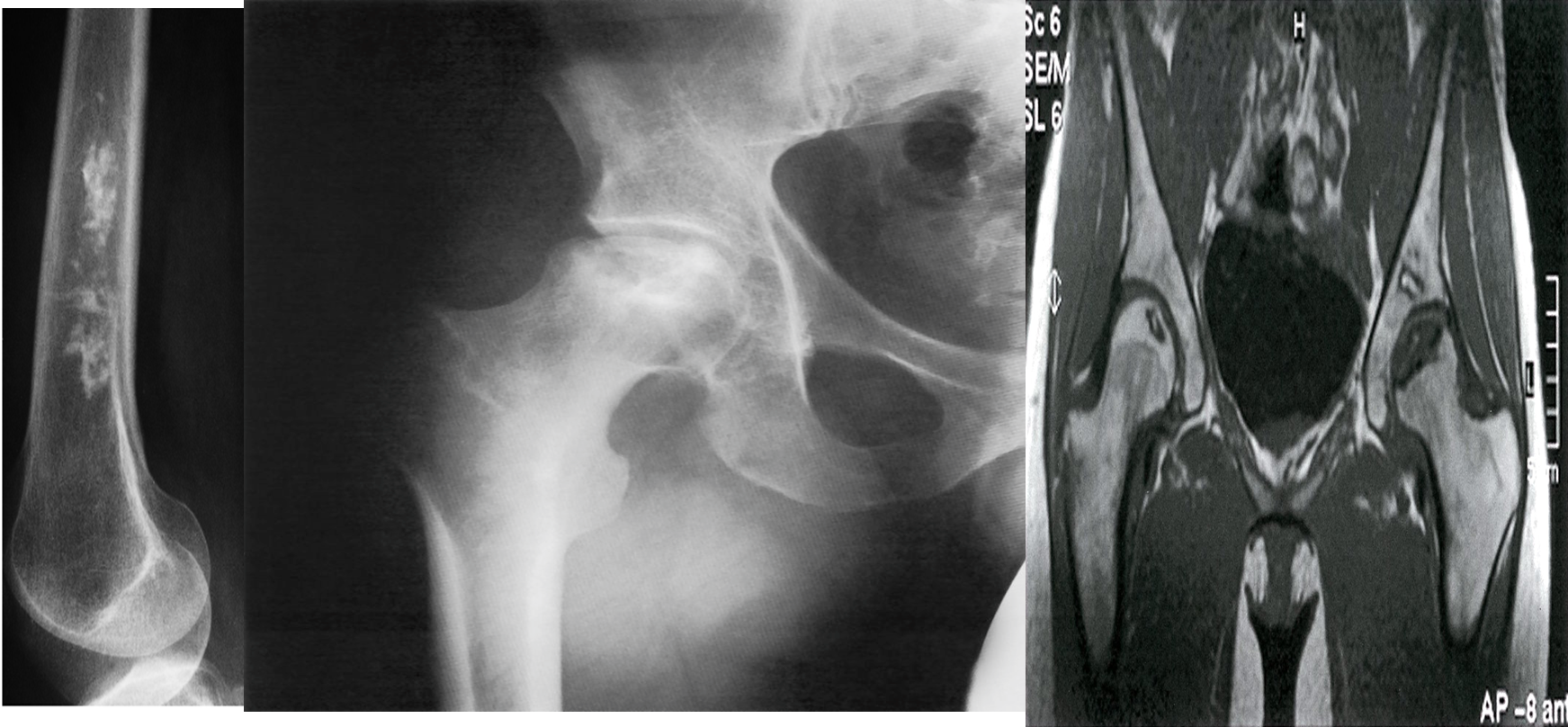 A) Bone infarct. There is calcification in the medulla of the lower end of the femur.
A) Bone infarct. There is calcification in the medulla of the lower end of the femur.
B) X-ray right hip (AP view) The articular surface of the femoral head is irregular (black dotted line) and the underlying bone is heterogeneous (green overlay) as a result of osteonecrosis. A zone of sclerosis (red overlay) is present along the interface with normal bone.
C) Bilateral osteonecrosis of the femoral head
MRI hips (T1 weighted; coronal view) of a 32-year-old woman with groin pain
There is a well-defined crescent-shaped area of intermediate to low signal within the subchondral marrow of the left femoral head . A thick peripheral band of even lower signal separates this region from the hyperintense normal bone marrow. On the right side, there is a slightly smaller area of abnormal oval-shaped signal within the epiphysis of the right femoral head.