Chromosomal Abnormalities and Mode of Inheritance of Single Gene Trait
Prepared by:
*Dr. Salma Elgazzar *
LEARNING OBJECTIVES
- Understand the principles of genetic inheritance and define single gene (autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive) and multifactorial inheritance and chromosomal disorders.
- List common prenatal diagnostic assessments (maternal serum screening, amniocentesis, chorionic villus sampling, and ultrasonography) and understand their use.
- Explain the use of family history to construct a pedigree in the evaluation of possible genetic disorders.
- List the indications for obtaining chromosome studies.
- Recognize the role of careful history taking and physical examination in the evaluation of a patient with structural or developmental abnormalities (e.g., facial features, palmar crease, measurements, symmetry).
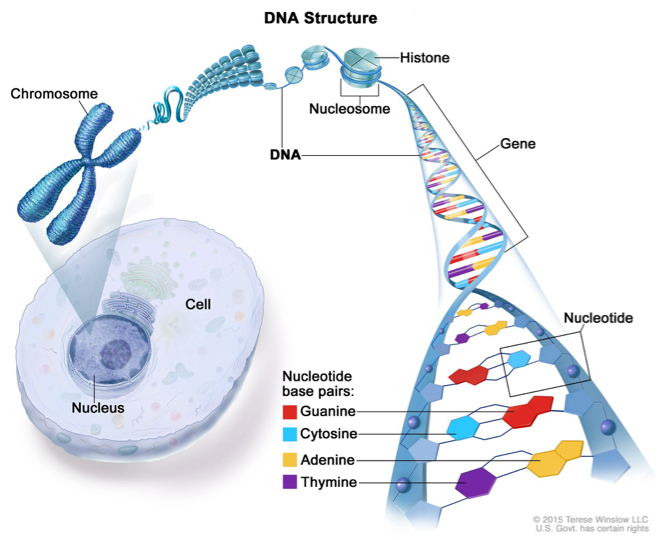
Chromosome Structure
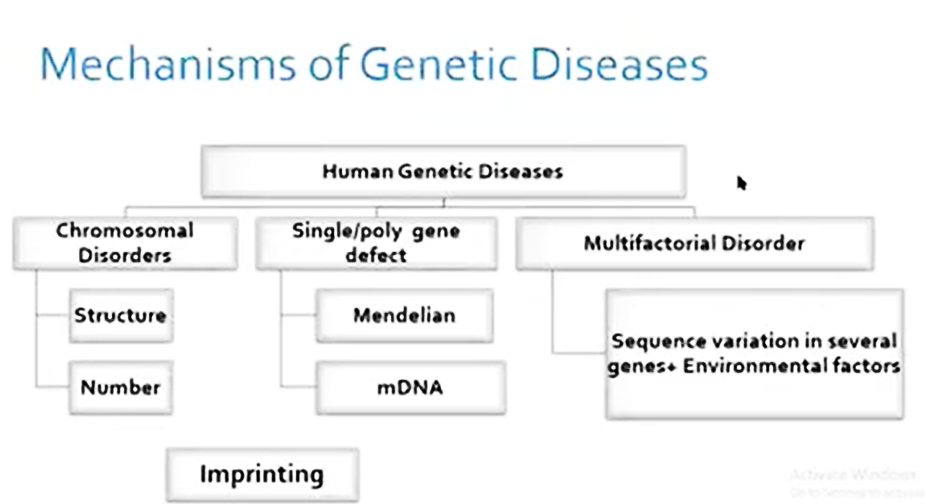
GENETIC DISORDERS
- Genetic disorders are congenital. Some genetic disorders also occur after birth or at any stage of life.
- The vast majority are incurable and have a complete phenotypic profile.
- The abnormality occurs mainly due to mutations in either gene or chromosome.
•Any alteration (deletion, addition, duplication or inversion) occurs in a gene or DNA sequence is known as gene mutation.
•Any alteration or variation (deletion, addition, duplication, invention or change in number) happens in a chromosome is known as chromosomal mutation.
•Besides these major two types of categories, some other type of mutations also occurs in a genome. e.g.triplet repeats expansion.
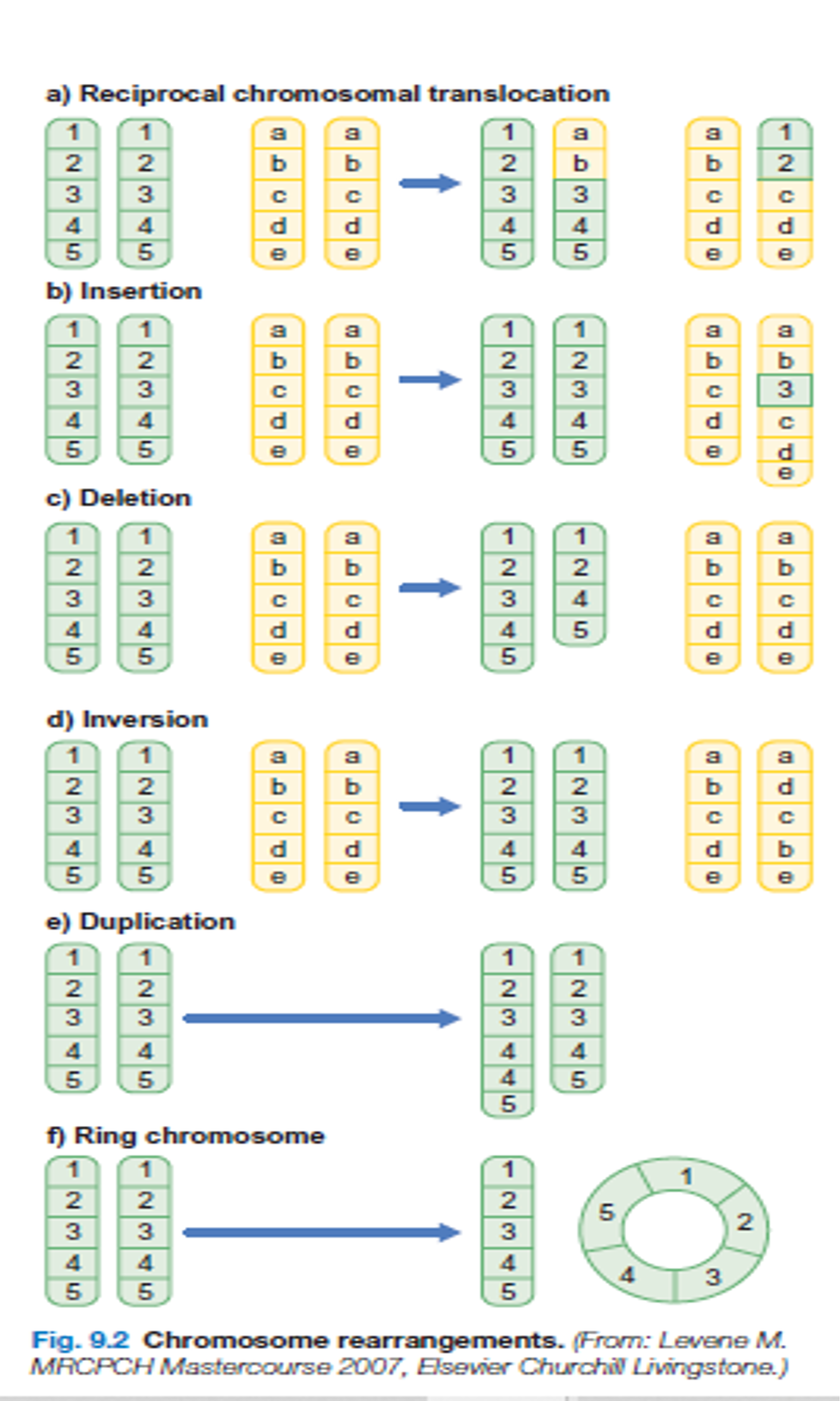
CLASSIFICATION OF CHROMOSOMAL ANOMALIES
Numerical (usually due to de novo error in meiosis)
- Aneuploidy
- monosomy
- trisomy
- Polyploidy
- triploidy
Structural (may be due to de novo error in meiosis or inherited)
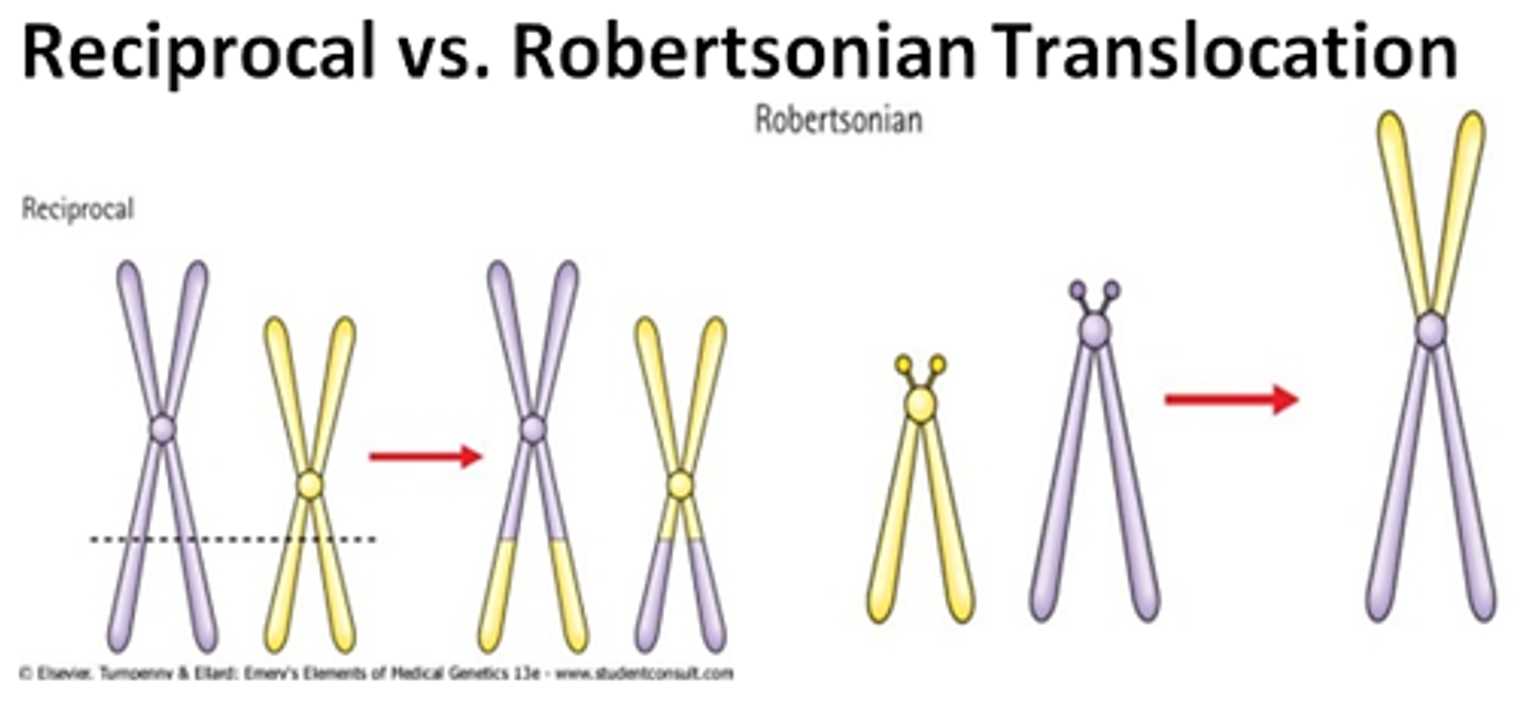
- Translocations
- reciprocal
- Robertsonian (centric fusion)
- Deletions
- Duplications
- Inversions
Different cell lines (occurs post-zygotically)
- Mosaicism
In some cases ,only a portion of cells that make up a person ‘s body are affected by a single –gene defect, a geneomic disorder ,or a chromosomal defect.this is referred to as mosaicism indicates that the individual’s body is made up of 2 or more distinct cell population
CLASSIFICATION OF GENE DEFECT ANOMALIES
Most heritable genetic (Mendelian) disorders are caused by mutations in a single gene from the 20,000 individual genes in the human genome. For many disorders, the Mendelian pattern of inheritance is known. A family tree (pedigree) is an essential part of genetic evaluation.
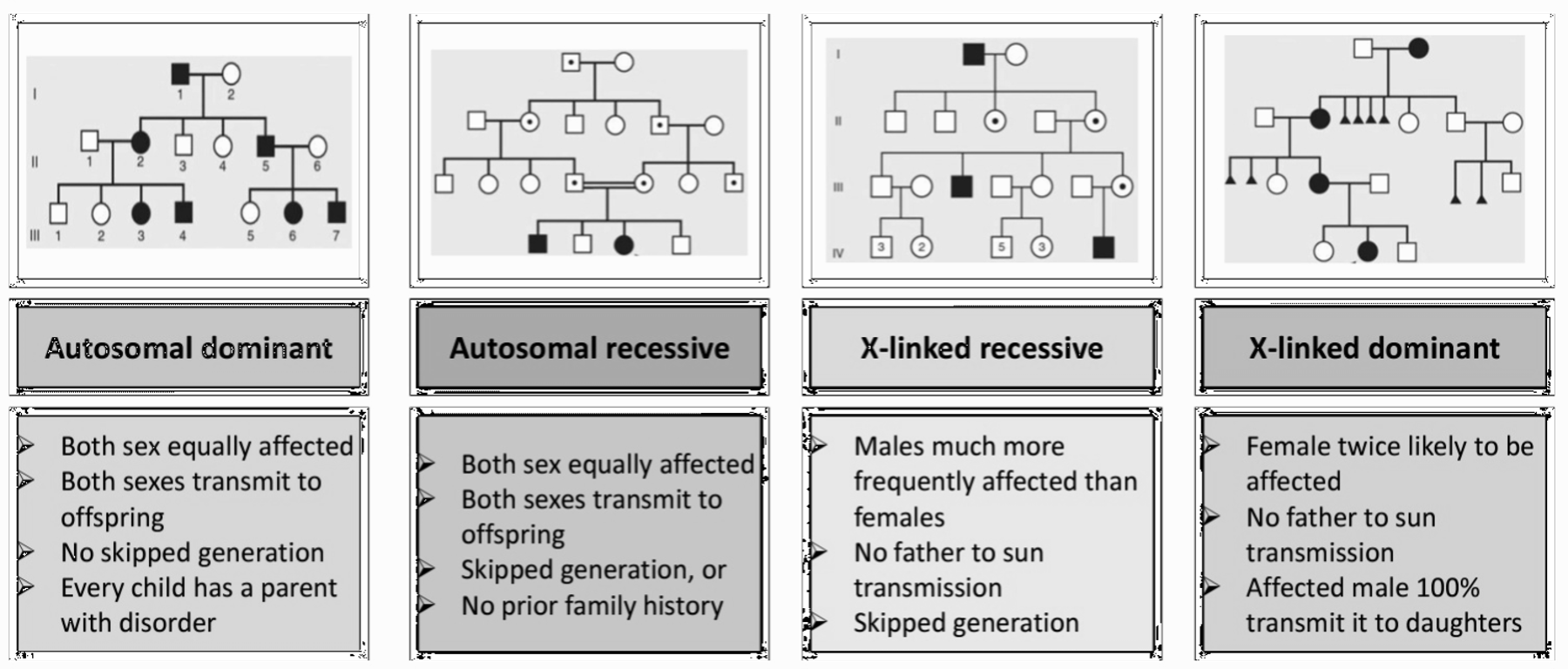
Types of Inheritance
Here’s a comprehensive table comparing the different modes of Mendelian inheritance based on the information provided:
| Inheritance Type | Key Characteristics | Chance of Inheritance | Affected Individuals | Examples of Disorders | Notes |
|---|---|---|---|---|---|
| Autosomal Dominant (AD) | - Affected individual carries the abnormal gene on one of a pair of autosomes. - Trait appears in every generation. - Males and females are equally affected. - Male-to-male transmission occurs. | - Each child of an affected parent has a 50% chance of inheriting the mutated gene. - Variation in expression, non-penetrance, new mutation, parental mosaicism, or homozygosity may occur. | - Affected individuals can have an affected parent or a new mutation. - Both sexes equally affected. | - Achondroplasia - Ehlers–Danlos syndrome - Familial hypercholesterolaemia - Marfan syndrome - Neurofibromatosis - Noonan syndrome - Osteogenesis imperfecta | - Traits generally involve mutations in genes coding for regulatory or structural proteins (e.g., collagen). |
| Autosomal Recessive (AR) | - Affected individuals are usually homozygous for the abnormal gene. - Each unaffected parent is a heterozygous carrier. - Risk varies between populations and is increased by consanguinity. | - If both parents are carriers, each child has a 25% chance of being affected. | - Affected children are usually born to unaffected parents who are carriers. | - Sickle cell disease - Thalassemia - Congenital adrenal hyperplasia - Cystic fibrosis - Friedreich ataxia - Galactosemia - Glycogen storage diseases - Hurler syndrome - Oculocutaneous albinism - Phenylketonuria | - Often affect metabolic pathways. |
| X-Linked Recessive | - Males are primarily affected. - Female carriers are usually healthy; occasionally show features of the disease. - Family history may be negative due to new mutations or gonadal mosaicism. | - Each son of a female carrier has a 50% risk of being affected. - Each daughter has a 50% risk of being a carrier. | - Daughters of affected males will all be carriers. - Sons of affected males will not be affected. | - Hemophilia A and B - Glucose-6-phosphate dehydrogenase deficiency - Color blindness - Duchenne muscular dystrophy - Fragile X syndrome | - Identifying female carriers is important for genetic counseling. |
| X-Linked Dominant | - Both males and females are affected. - Females carrying the mutation are affected; males have a more severe condition. | - Not specified, but typically involves affected females passing the mutation to offspring. | - Males with the mutation may have lethal conditions. - Females may have milder symptoms. | - Hypophosphataemic (vitamin D-resistant) rickets - Rett syndrome (in females) | - Unusual inheritance pattern compared to other types. |
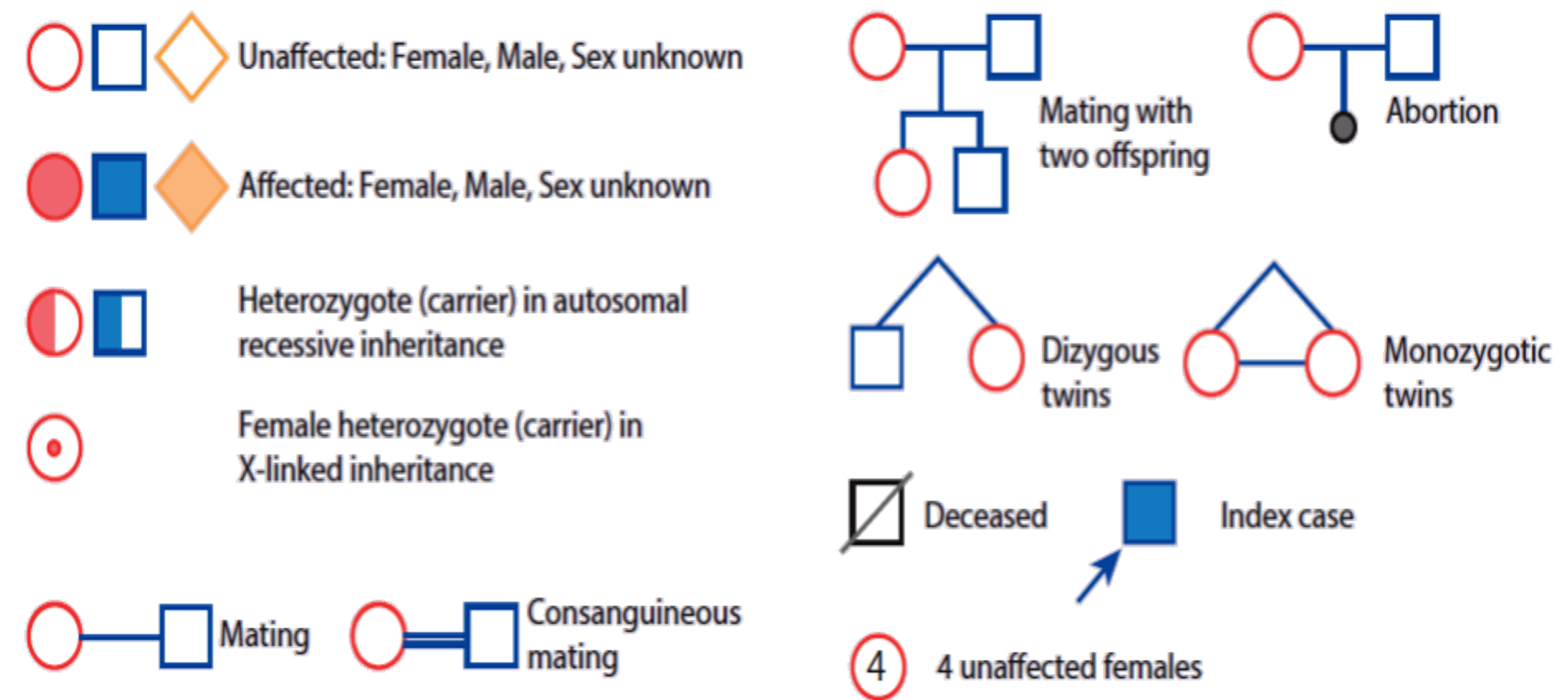
PEDIGREE Z OSPE
When constructing a pedigree, ask about miscarriages, stillbirths, and consanguinity. Take details from both sides of the family and record dates of birth rather than current ages.
POLYGENIC, MULTIFACTORIAL OR COMPLEX INHERITANCE
Many factors are involved in causing a birth defect.
- A combination of genes from both parents, in addition to environmental factors, produces the trait or condition.
- Often one gender (either males or females) is affected more frequently than the other in multifactorial traits. For example, hip dysplasia is nine times more common in females than males.
Multifactorial inheritance characteristics:
- The higher the number of affected individuals in the family, the higher the recurrence risks.
- The recurrence risk is higher if the affected individual is a member of the less commonly affected sex (e.g., Autism is more common in boys than girls, but if a girl in the family has autism, it is twice as likely to recur in a sibling than if a boy is the one with autism).
- Recurrence risk is higher if the affected individual suffers the more severe form of the disease.
- Recurrence risk correlates with the prevalence in the general population.
| Category | Examples |
|---|---|
| Congenital abnormalities | - Cleft lip and palate - Neural tube defect (environmental such folic) - Pyloric stenosis (first baby male commonly) |
| Immune mediated disorders | - Atopy - Inflammatory bowel disease - Diabetes mellitus type 1 - Juvenile idiopathic arthritis |
| Metabolic disorders | - Obesity - Diabetes mellitus type 2 |
| Neurological disorders | - Epilepsy - Autism - Attention deficit disorder |
FAMILY HISTORY & PEDIGREE IN THE EVALUATION OF POSSIBLE GENETIC DISORDERS
Use of family history to assess genetic risk: Key factors that suggest the presence of a genetic disorder include the following:
- Multiple affected individuals in multiple generations from either side of the individual’s family.
- Disease occurrence at an earlier age than usual.
- Close degree of relatedness (i.e., first- or second-degree relative) between affected relatives and the individual.
- Presence of associated conditions in the family.
- Presence of consanguinity.
- Multiple miscarriages.
- Congenital anomalies.
Family/Medical History from the Referring Clinician
-
Collection of potentially relevant information limited to
- first-degree relatives (parents, siblings, children) and
- second-degree relatives (e.g., grandparents, aunts, uncles, grandchildren, half-siblings);
- third-degree relatives may also be included.
-
Patient concerns about disease, problems with pregnancy, occurrence of congenital anomalies, early deaths or disease onset, and determination of non-genetic risk factors for disease.
-
Assessment of consanguinity, in which parents share a common ancestor, can also be helpful for determining the risk for an autosomal recessive condition.
-
Precise medical diagnoses in family members can provide revealing information regarding symptoms, surgeries, and medication use.
-
The medical conditions of siblings or other relatives who have died (often the most informative information).
•The history taken should include medical and obstetric history, such as the duration of gestation, prenatal care, and maternal exposures (e.g., alcohol, prescribed or illicit drugs, cigarettes, fevers, illnesses, chemicals, radiation).
• A history of stillbirths and miscarriages .
•Results of noninvasive and invasive prenatal testing, including ultrasound examinations, should be obtained.
•A complete family history and pedigree (ideally four generations) should be obtained to identify medical problems that may be related to the birth defect or other medical issues that can impact the family (i.e., multiple cancers).
•The age of the parents is important because the incidence of chromosome aneuploidies is increased in older mothers and denovo autosomal-dominant pathogenic variants (eg, achondroplasia, neurofibromatosis type 1) are more commonly seen with older parents, with the effect being more marked for advanced paternal age. consanguinity may suggest the possibility of autosomal-recessive disorders.
•Determining ethnic/racial origin may be helpful.
COMMON PRENATAL DIAGNOSTIC ASSESSMENT
The main diagnostic techniques for antenatal diagnosis:
-
Maternal serum screening: e.g., maternal serum alpha-fetoprotein
- Which is high in neural tube defects (spina bifida),
- low in trisomies like Down syndrome.
-
Detailed ultrasound scanning: Fetal growth, structural malformation, amniotic fluid volume.
-
Fetal blood sampling.
-
Amniocentesis.
-
Non-invasive prenatal testing.
PHYSICAL EXAMINATION
A thorough physical examination should be performed. The standard measurements: head circumference (occipital frontal circumference), height/length, and weight. Arm span and lower segment/upper segment.
Measurements of craniofacial structures: ear length, inner canthal distance, outer canthal distance, interpupillary distance, palpebral fissures length, and philtrum length (extends from the nasal septum to the upper vermilion border).
Length of hands, feet, and digits to look for brachydactyly or arachnodactyly.
In newborns and fetuses, the placenta and umbilical cord should also be examined. Two-vessel cord is associated with congenital heart defects.
Examination of family members may assist with the evaluation of some abnormalities. One example is Treacher-Collins syndrome, an autosomal-dominant disorder in which a parent can have minimal microtia or mild underdevelopment of malar facial structures that may have been missed on previous examinations.
The Physical Exam is a Powerful Tool for the Geneticist and the Clinician
Chromosomal Abnormality Examination
CHROMOSOMAL STUDIES / INVESTIGATION
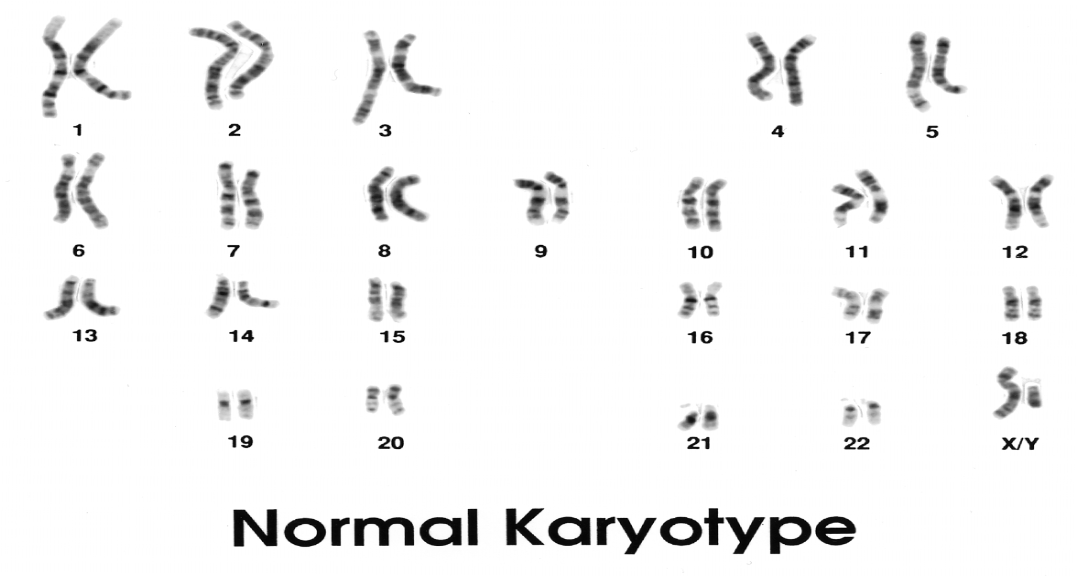
Karyotyping:
A karyotype is a photomicrograph of an individual’s chromosomes arranged in a standard format showing the number, size, and shape of each chromosome.
The karyotype can identify abnormalities of the number or structure of the chromosomes. A karyotype may be done on white blood cells, amniocytes, or skin fibroblasts.
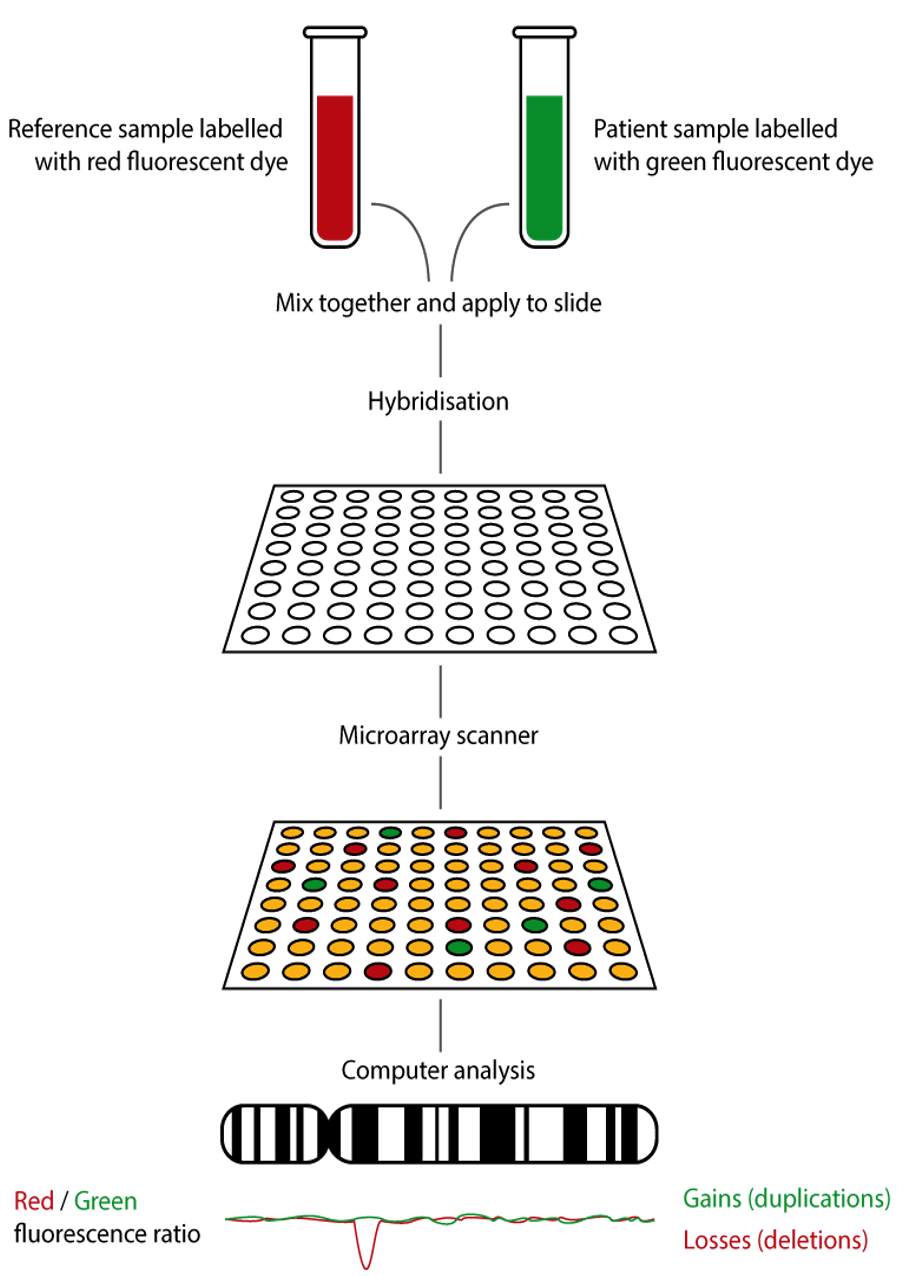
Array Comparative Genomic Hybridization (aCGH) Array CGH, or microarray, is a relatively new technique that looks for chromosomal copy number variations (CNVs) by comparing a patient’s DNA to normal control DNA.
It has taken over from karyotype as the first-line test for most chromosome abnormalities in pediatric practice.
In CGH, the patient DNA is labelled green and the normal control DNA is labelled red. Thousands of different probes (fragments of DNA) which bind specifically to regions spanning the whole genome are immobilized on a slide, the array.
Array CGH is typically able to detect deletions and duplications larger than approximately 50,000 bases.
This is a resolution more than 100 times higher than karyotyping and allows the detection of microdeletions or duplications as well as other larger chromosome copy number abnormalities, including aneuploidies.
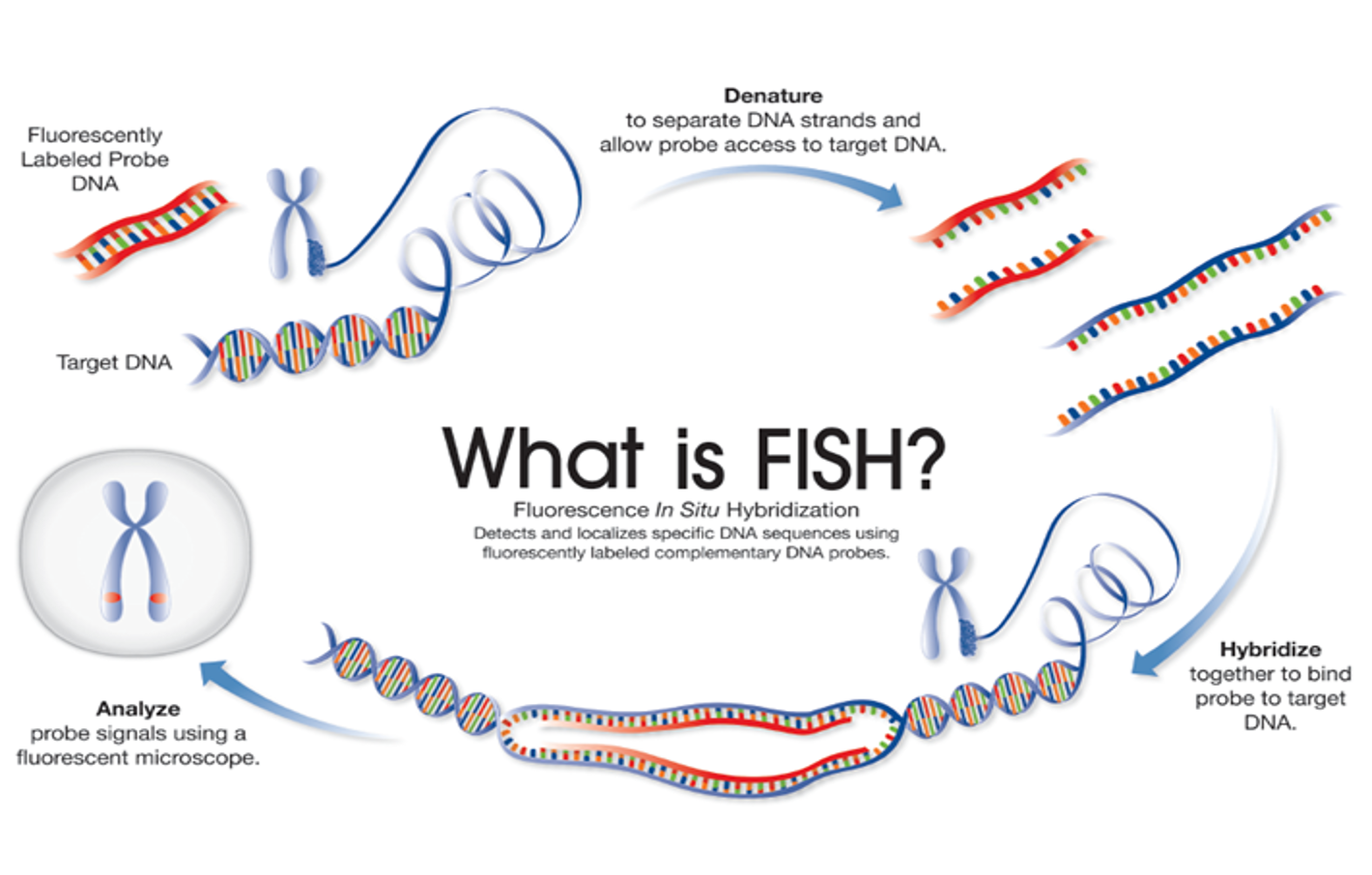
Fluorescence In Situ Hybridization (FISH) FISH is a technique that uses fluorescently labelled DNA sequences (probes) to assess specific regions of interest, such as genes and centromeres using fluorescent microscopy.
The DNA sequence of the FISH probe is complementary to the region of interest.
When using a FISH probe, if the region of interest is present, the probe will bind (hybridize) to the target DNA sequence, which can then be visualized using fluorescent microscopy – indicating the presence of the gene/centromere, etc.
If the region of interest has been deleted, the probe will not bind to the target DNA and so will not be visualized.